
Selective Sweeps and Polygenic Adaptation Drive Local Adaptation along Moisture and Temperature Gradients in Natural Populations of Coast Redwood and Giant Sequoia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Sequencing, and SNP Calling
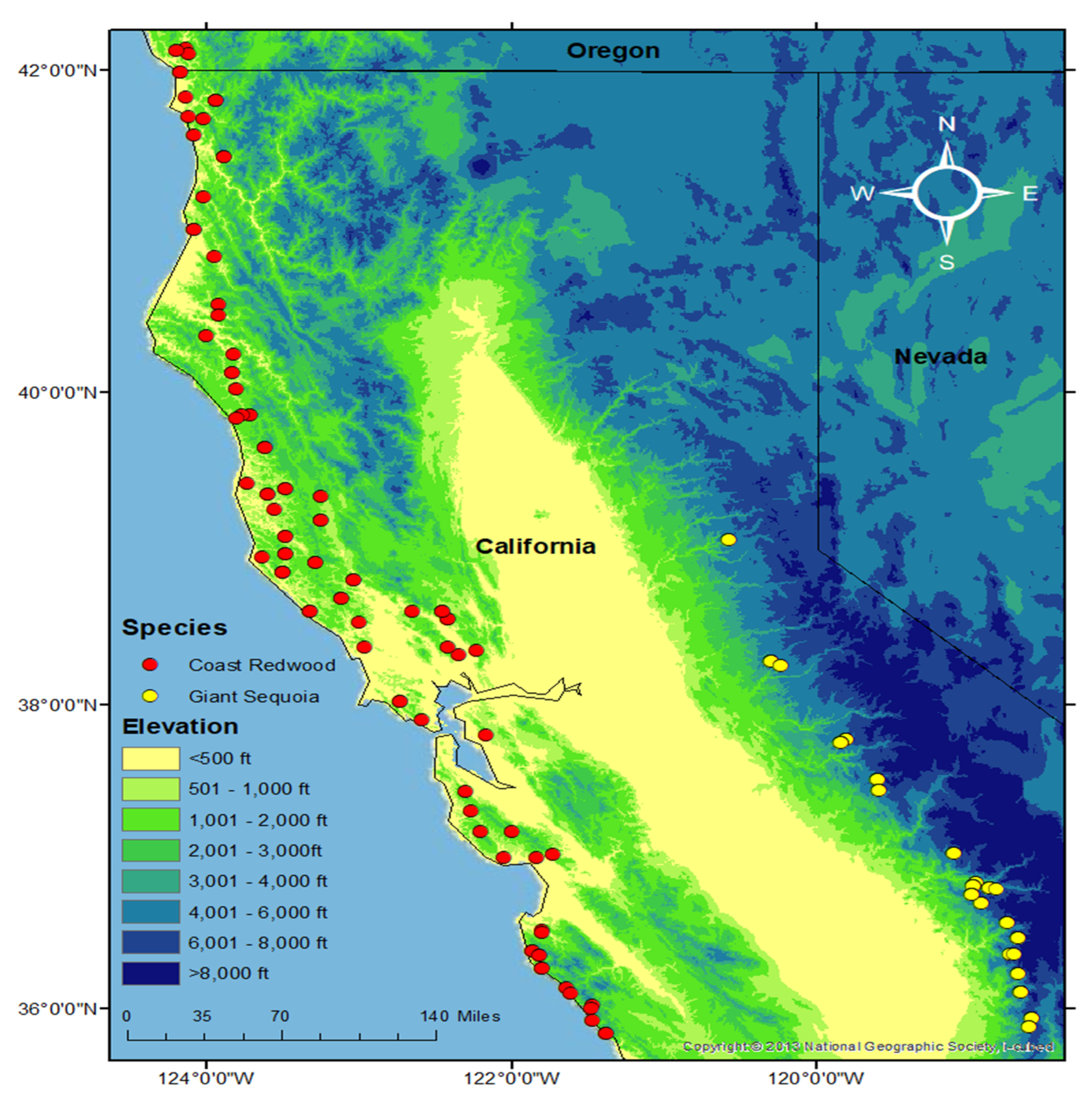
2.2. Population Structure
2.3. Genome-Wide Patterns of Diversity and Differentiation
2.4. Signatures of Positive Selection in Outlier Regions
2.5. Linkage Disequilibrium (LD) Analysis
2.6. Genome-Wide Environmental Association (GEA)
2.7. Functional Gene Annotations and Enrichment Analyses
3. Results
3.1. Sequence Capture and SNP Datasets
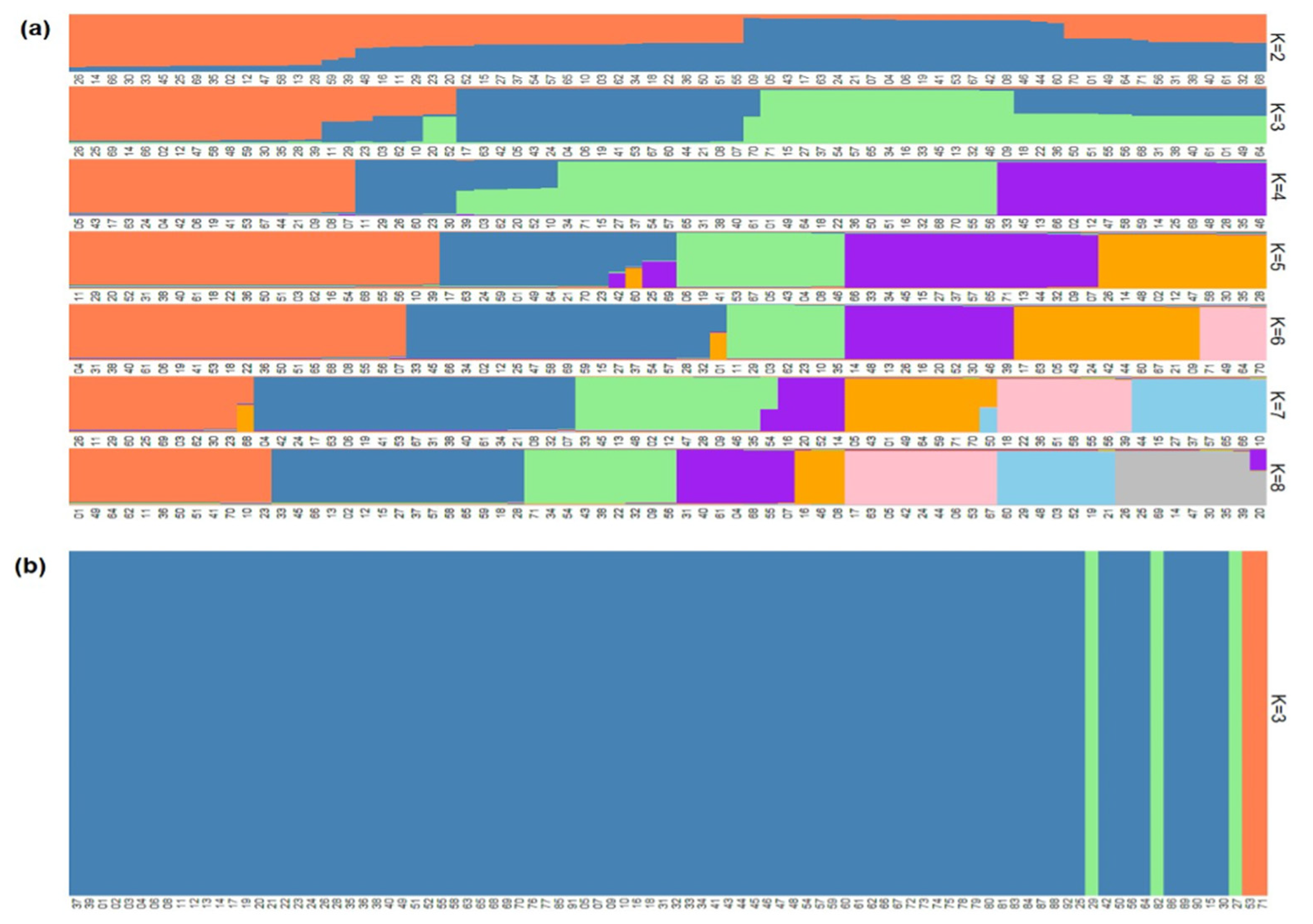
3.2. Population Structure
3.3. Genome-Wide Patterns of Diversity and Differentiation
3.4. Signatures of Positive Selection in Outlier Regions
3.5. Genome-Wide Linkage Disequilibrium (LD)
3.6. Genome-Wide Environmental Association (GEA)
3.7. Gene Enrichment Analyses
4. Discussion
4.1. Selective Sweeps and Polygenic Adaptation Drive Genomic Architecture in the Species
4.2. Patterns of Genomic Diversity and Divergence
4.3. Moisture-Related Variables Drive Adaptation in the Species
4.4. Functional Annotation of Genes Associated with Environmental Variables
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stetter, M.G.; Thornton, K.; Ross-Ibarra, J. Genetic architecture and selective sweeps after polygenic adaptation to distant trait optima. PLoS Genet. 2018, 14, e1007794. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Pickrell, J.K.; Coop, G. The genetics of human adaptation: Hard sweeps, soft sweeps, and polygenic adaptation. Curr. Biol. 2010, 20, R208–R215. [Google Scholar] [CrossRef] [Green Version]
- Chevin, L.M.; Hospital, F. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics 2008, 180, 1645–1660. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.; Clark, A.; Bustamante, C.D. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.T.; Lee, C.R.; Rushworth, C.A.; Colautti, R.I.; Mitchell-Olds, T. Genetic tradeoff and conditional neutrality contribute to local adaptation. Mol. Ecol. 2013, 22, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, N.L.; Hudson, R.R.; Langley, C.H. The ‘hitchhiking effect’ revisited. Genetics 1989, 123, 887–899. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef] [Green Version]
- Hermisson, J.; Pennings, P.S. Soft sweeps: Molecular population genetics of adaptation from standing genetic variation. Genetics 2005, 169, 2335–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przeworski, M.; Coop, G.; Wall, J.D. The signature of positive selection on standing genetic variation. Evolution 2005, 59, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, J.; Pennings, P.S. Soft sweeps and beyond: Understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods Ecol. Evol. 2017, 8, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.; Stephan, W. Modes of rapid polygenic adaptation. Mol. Biol. Evol. 2017, 34, 3169–3175. [Google Scholar] [CrossRef]
- Jain, K.; Stephan, W. Rapid adaptation of a polygenic trait after a sudden environmental shift. Genetics 2017, 206, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Di Rienzo, A. Adaptation–not by sweeps alone. Nat. Rev. Genet. 2010, 11, 665–667. [Google Scholar] [CrossRef] [Green Version]
- Barghi, N.; Schlotterer, C. Distinct patterns of selective sweep and polygenic adaptation in evolve and resequence studies. Genome Biol. Evol. 2020, 12, 890–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollstein, A.; Stephan, W. Adaptive fixation in two-locus models of stabilizing selection and genetic drift. Genetics 2014, 198, 685–697. [Google Scholar] [CrossRef]
- Pavlidis, P.; Jensen, J.D.; Stephan, W.; Stamatakis, A. A critical assessment of storytelling: Gene ontology categories and the importance of validating genomic scans. Mol. Biol. Evol. 2012, 29, 3237–3248. [Google Scholar] [CrossRef] [Green Version]
- Sanjak, J.S.; Sidorenko, J.; Robinson, M.R.; Thornton, K.R.; Visscher, P.M. Contemporary directional and stabilizing selection. Proc. Natl. Acad. Sci. USA 2018, 115, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Torre, A.R.; Wilhite, B.; Neale, D.B. Environmental genome-wide association reveals climate adaptation is shaped by subtle to moderate allele frequency shifts in loblolly pine. Genome Biol. Evol. 2019, 11, 2976–2989. [Google Scholar] [CrossRef] [PubMed]
- Forester, B.R.; Lasky, J.R.; Wagner, H.H.; Urban, D.L. Comparing methods for detecting multilocus adaptation with multivariate genotype-environment associations. Mol. Ecol. 2018, 27, 2215–2233. [Google Scholar] [CrossRef]
- De Kort, H.; Vandepitte, K.; Bruun, H.H.; Closset-Kopp, D.; Honnay, O.; Mergeay, J. Landscape genomics and a common garden trial reveal adaptive differentiation to temperature across Europe in the tree species Alnus glutinosa. Mol. Ecol. 2014, 23, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Hancock, A.M.; Alkorta-Aranburu, G.; Witonsky, D.B.; DiRienzo, A. Adaptations to new environments in humans: The role of subtle allele frequency shifts. Philos. Trans. R. Soc. B 2010, 365, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.R.; Des Marais, D.L.; McKAY, J.K.; Richards, J.H.; Juenger, T.E.; Keitt, T.H. Characterizing genomic variation of Arabidopsis thaliana: The roles of geography and climate. Mol. Ecol. 2012, 21, 5512–5529. [Google Scholar] [CrossRef] [PubMed]
- Steane, D.A.; Potts, B.M.; McLean, E.; Prober, S.M.; Stock, W.D.; Vaillancourt, R.E.; Byrne, M. Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol. Ecol. 2014, 23, 2500–2513. [Google Scholar] [CrossRef]
- Mitton, J.B.; Linhart, Y.B.; Hamrick, J.L.; Beckman, J.S. Observations on the genetic structure and mating system of ponderosa pine in the Colorado front range. Theor. Appl. Genet. 1977, 51, 5–13. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into Conifer Giga-genomes. Plant Physiol. 2014, 166, 1724–1732. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.; Doerksen, T.; Boyle, B.; Clément, S.; Deslauriers, M.; Beauseigle, S.; Blais, S.; Poulin, P.; Lenz, P.; Caron, S.; et al. Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 2011, 188, 197–214. [Google Scholar] [CrossRef] [Green Version]
- Cumbie, W.P.; Eckert, A.J.; Wegrzyn, J.; Whetten, R.W.; Neale, D.B.; Goldfarb, B. Association genetics of carbon isotope discrimination, height and foliar nitrogen in a natural population of Pinus taeda L. Heredity 2011, 107, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Martinez, S.C.; Huber, D.; Ersoz, E.; Davis, J.M.; Neale, D.B. Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity 2008, 101, 19–26. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Wilhite, B.; Puiu, D.; St Clair, J.B.; Crepeau, M.W.; Salzberg, S.L.; Langley, C.H.; Allen, B.; Neale, D.B. Dissecting the polygenic basis of cold adaptation using genome-wide association of traits and environmental data in Douglas-fir. Genes 2021, 12, 110. [Google Scholar] [CrossRef]
- Lu, M.; Krutovsky, K.; Nelson, C.D.; West, J.B.; Reilly, N.A.; Loopstra, C.A. Association genetics of growth and adaptive traits in loblolly pine (Pinus taeda L.) using whole-exome-discovered polymorphisms. Tree Genet. Genomes 2017, 13, 57–75. [Google Scholar] [CrossRef]
- Lu, M.; Seeve, C.M.; Loopstra, C.A.; Krutovsky, K.V. Exploring the genetic basis of gene transcript abundance and metabolite levels in loblolly pine (Pinus taeda L.) using association mapping and network construction. BMC Genet. 2018, 19, 100–113. [Google Scholar] [CrossRef]
- Yeaman, S.; Hodgins, K.A.; Lotterhos, K.E.; Suren, H.; Nadeau, S.; Degner, J.C.; Nurkowski, K.A.; Smets, P.; Wang, T.; Gray, L.K.; et al. Convergent local adaptation to climate in distantly related conifers. Science 2016, 353, 1431–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, S. A record of Sequoia from the Jurassic of Manchuria. Bot. Gaz. 1951, 113, 228–230. [Google Scholar]
- Miller, C.N. Mesozoic conifers. Bot. Rev. 1977, 43, 217–280. [Google Scholar] [CrossRef]
- Lanner, R.M. Conifers of California; Cachuma Press: Los Olivos, CA, USA, 1999; ISBN 0-9628505-9628503-9628505. [Google Scholar]
- Sillett, S.C.; Van Pelt, R.; Carroll, A.L.; Kramer, R.D.; Ambrose, A.R.; Trask, D. How do tree structure and old age affect growth potential of California redwoods? Ecol. Monogr. 2015, 85, 181–212. [Google Scholar] [CrossRef]
- Scott, A.D.; Zimin, A.V.; Puiu, D.; Workman, R.; Britton, M.; Zaman, S.; Caballero, M.; Read, A.C.; Bogdanove, A.J.; Burns, E.; et al. A reference genome sequence for Giant Sequoia. G3 (Bethesda) 2020, 10, 3907–3919. [Google Scholar] [CrossRef] [PubMed]
- Weatherspoon, C.P. Sequoiadendron giganteum (Lindl.) Buchholz Giant Sequoia. 1990. Available online: https://dendro.cnre.vt.edu/dendrology/USDAFSSilvics/136.pdf (accessed on 1 March 2021).
- Dodd, R.S.; DeSilva, R. Long-term demographic decline and late glacial divergence in a Californian paleoendemic: Sequoiadendron giganteum (giant sequoia). Ecol. Evol. 2016, 6, 3342–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, H.R.; Azuma, W.; Kuroda, K.; Sillett, S.C. Pushing the limits to tree height: Could foliar water storage compensate for hydraulic constraints in Sequoia sempervirens? Funct. Ecol. 2014, 28, 1087–1093. [Google Scholar] [CrossRef]
- Neale, D.B.; Zimin, A.V.; Zaman, S.; Scott, A.D.; Shrestha, B.; Workman, R.E.; Puiu, D.; Allen, B.J.; Sekhwal, M.K.; De La Torre, A.R.; et al. Assembled and annotated 26.5 Gbp coast redwood genome: A resource for estimating evolutionary adaptive potential and investigating hexaploidy origin. G3 Genes Genomes Genet. 2021, in press. [Google Scholar]
- Ahuja, M.R. Genetic constitution and diversity in four narrow endemic redwoods from the family Cupressaceae. In Biodiversity and Conservation of Woody Plants; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; Volume 165, pp. 5–19. [Google Scholar]
- Burns, E.E.; Campbell, R.; Cowan, P.D. State of Redwoods Conservation Report: A Tale of Two Forests, Coast Redwoods, Giant Sequoia; Save the Redwoods League: San Francisco, CA, USA, 2018. [Google Scholar]
- Farjon, A.; Schmid, R. Sequoia sempervirens. The IUCN red list of threatened species 2013, e.T34051A2841558. Available online: https://www.iucnredlist.org/species/34051/2841558 (accessed on 1 April 2021).
- Breidenbach, N.; Gailing, O.; Krutovsky, K.V. Genetic structure of coast redwood (Sequoia sempervirens [D. Don] Endl.) populations in and outside of the natural distribution range based on nuclear and chloroplast microsatellite markers. PLoS ONE 2020, 15, e0243556. [Google Scholar] [CrossRef] [PubMed]
- Breidenbach, N.; Sharov, V.V.; Gailing, O.; Krutovsky, K.V. De novo transcriptome assembly of cold stressed clones of the hexaploid Sequoia sempervirens (D. Don) Endl. Sci. Data 2020, 7, 239–247. [Google Scholar] [CrossRef]
- DeSilva, R.; Dodd, R.S. Association of genetic and climatic variability in giant sequoia, Sequoiadendron giganteum, reveals signatures of local adaptation along moisture-related gradients. Ecol. Evol. 2020, 10, 10619–10632. [Google Scholar] [CrossRef]
- Klápště, J.; Meason, D.; Dungey, H.S.; Telfer, E.J.; Silcock, P.; Rapley, S. Genotype-by-environment interaction in coast redwood outside natural distribution search for environmental cues. BMC Genet. 2020, 21, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.D.; Stenz, N.W.; Ingvarsson, P.K.; Baum, D.A. Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers. New Phytol. 2016, 211, 186–193. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Sekhwal, M.K.; Puiu, D.; Salzberg, S.L.; Scott, A.D.; Allen, B.; Neale, D.B.; Chin, A.R.O.; Buckley, T.N. Genome-wide association identifies candidate genes for drought tolerance in coast redwood and giant sequoia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.-Y.; Myles, S. LinkImpute: Fast and accurate genotype imputation for nonmodel organisms. G3 (Bethesda) 2015, 5, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.D.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Ke, X.; Hunt, S.; Tapper, W.; Lawrence, R.; Stavrides, G.; Ghori, J.; Whittaker, P.; Collins, A.; Morris, A.P.; Bentley, D.; et al. The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum. Mol. Genet. 2004, 13, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.G.; Weir, B.S. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genet. Res. 2011, 93, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Wang, T.L.; Hamann, A.; Spittlehouse, D.; Carroll, C. Locally downscaled and spatially customizable climate data for historical and future periods for North America. PLoS ONE 2016, 11, e0156720. [Google Scholar] [CrossRef]
- Caye, K.; Jumentier, B.; Lepeule, J.; Francois, O. LFMM 2: Fast and accurate inference of gene-environment associations in genome-wide studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; Francois, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 7, 1687–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R. Vegan: Community Ecology Package; CRAN: Helsinki, Finland, 2020; Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 2 February 2021).
- Dormann, C.F.; Elith, J.; Bacher, S.; Buchmann, C.; Carl, G.; Carré, G.; Marquéz, J.R.G.; Gruber, B.; Lafourcade, B.; Leitão, P.J.; et al. Collinearity: A review of methods to deal with it and a simulation study evaluating their performance. Ecography 2013, 36, 27–46. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Hayward, L.K.; Sella, G. Polygenic adaptation after a sudden change in environment. bioRxiv 2021, 792952. [Google Scholar] [CrossRef]
- Chhatre, V.E.; Fetter, K.C.; Gougherty, A.V.; Fitzpatrick, M.C.; Soolanayakanahally, R.Y.; Zalensy, R.S. Climatic niche predicts the landscape structure of locally adaptive standing genetic variation. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- De La Torre, A.R.; Li, Z.; Van de Peer, Y.; Ingvarsson, P.K. Contrasting rates of molecular evolution and patterns of selection among gymnosperms and flowering plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef] [Green Version]
- Neale, D.B.; Savolainen, O. Association genetics of complex traits in conifers. Trends Plant. Sci. 2004, 9, 325–330. [Google Scholar] [CrossRef]
- Namroud, M.C.; Guillet-Claude, C.; Mackay, J.; Isabel, N.; Bousquet, J. Molecular evolution of regulatory genes in spruces from different species and continents: Heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J. Mol. Evol. 2010, 70, 371–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutovsky, K.V.; Neale, D.B. Nucleotide diversity and linkage disequilibrium in cold-hardiness- and wood quality-related candidate genes in Douglas fir. Genetics 2005, 171, 2029–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birky, C.W. Heterozygosity, heteromorphy, and phylogenetic trees in asexual eukaryotes. Genetics 1996, 144, 427–437. [Google Scholar] [CrossRef]
- Peck, J.R.; Waxman, D. What’s wrong with a little sex? J. Evol. Biol. 2000, 13, 63–69. [Google Scholar] [CrossRef]
- Lotterhos, K.E.; Yeaman, S.; Degner, J.; Aitken, S.; Hodgins, K.A. Modularity of genes involved in local adaptation to climate despite physical linkage. Genome Biol. 2018, 19, 157–181. [Google Scholar] [CrossRef] [Green Version]
- Roschanski, A.M.; Csilléry, K.; Liepelt, S.; Oddou-Muratorio, S.; Ziegenhagen, B.; Huard, F.; Ullrich, K.K.; Postolache, D.; Vendramin, G.G.; Fady, B. Evidence of divergent selection for drought and cold tolerance at landscape and local scales in Abies alba Mill. in the French Mediterranean Alps. Mol. Ecol. 2016, 25, 776–794. [Google Scholar] [CrossRef]
- Mosca, E.; Gugerli, F.; Eckert, A.J.; Neale, D.B. Signatures of natural selection on Pinus cembra and P. mugo along elevational gradients in the Alps. Tree Genet. Genomes 2016, 12, 9–24. [Google Scholar] [CrossRef]
- Steane, D.A.; Potts, B.; McLean, E.H.; Collins, L.; Holland, B.; Prober, S.; Stock, W.D.; Vaillancourt, R.; Byrne, M. Genomic Scans across Three Eucalypts Suggest that Adaptation to Aridity is a Genome-Wide Phenomenon. Genome Biol. Evol. 2017, 9, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.M.; Slavov, G.T.; Rodgers-Melnick, E.; Martin, J.; Ranjan, P.; Muchero, W.; Brunner, A.M.; Schackwitz, W.; Gunter, L.; Chen, J.-G.; et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat. Genet. 2014, 46, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraldes, A.J.M.; Farzaneh, N.; Grassa, C.; McKown, A.; Guy, R.D.; Mansfied, S.D.; Douglas, C.J.; Cronk, Q.C.B. Landscape genomics of Populus trichocarpa: The role of hybridization, limited gene flow, and natural selection in shaping patterns of population structure. Evolution 2014, 68, 3260–3280. [Google Scholar] [CrossRef]
- Kijowska-Oberc, J.; Staszak, A.M.; Kaminski, J.; Ratajczak, E. Adaptation of forest trees to rapidly changing climate. Forests 2020, 11, 123. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, A.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant. Signal. Behav. 2011, 6, 1720–1731. [Google Scholar]
- Bathe, U.; Tissier, A. Cytochrome P450 enzymes: A driving force of plant diterpene diversity. Phytochemistry 2019, 161, 149–162. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, H.; Tong, X.; Liu, Z.; Zhang, X.; Li, D.; Jiang, X.; Yu, X. Identification and analysis of CYP450 and UGT supergene family members from the transcriptome of Aralia elata (Miq.) seem reveal candidate genes for triterpenoid saponin biosynthesis. BMC Plant Biol. 2020, 20, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Ran, F.; Dong, H.-L.; Wen, J.; Li, J.-N.; Liang, Z. Genome-wide analysis, classification, evolution, and expression analysis of the cytochrome P450 93 family in land plants. PLoS ONE 2016, 11, e0165020. [Google Scholar]
- Overmyer, K.; Vuorinen, K.; Brosche, M. Interaction points in plant stress signaling pathways. Physiol. Plant. 2018, 162, 191–204. [Google Scholar] [CrossRef]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of ubiquitin-mediated degradation system in plant biology. Front. Plant Sci. 2016, 7, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.Q.; Xue, H.W. The ubiquitin-proteasome system in plant responses to environments. Plant. Cell Environ. 2019, 42, 2931–2944. [Google Scholar] [PubMed] [Green Version]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; Campos, L.; Rivas, S. Roles of E3 ubiquitin-ligases in nuclear protein homeostasis during plant stress responses. Front. Plant Sci. 2018, 9, 139–146. [Google Scholar] [CrossRef]
- Mazzucotelli, E.; Belloni, S.; Marone, D.; De Leonardis, A.; Guerra, D.; Di Fonzo, N.; Cattivelli, L.; Mastrangelo, A. The e3 ubiquitin ligase gene family in plants: Regulation by degradation. Curr. Genom. 2006, 7, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Xue, B.; Xia, X.L.; Yin, W.L. A novel calcium-dependent protein kinase gene from Populus euphratica, confers both drought and cold stress tolerance. Biochem. Biophys. Res. Commun. 2013, 441, 630–636. [Google Scholar] [CrossRef]
- Hamel, L.P.; Miles, G.P.; Samuel, M.A.; Ellis, B.E.; Séguin, A.; Beaudoin, N. Activation of stress-responsive mitogen-activated protein kinase pathways in hybrid poplar (Populus trichocarpa x Populus deltoides). Tree Physiol. 2005, 25, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Su, H.; Han, L.; Wang, C.; Sun, Y.; Liu, F. Differential expression profiles of poplar MAP kinase kinases in response to abiotic stresses and plant hormones, and overexpression of PtMKK4 improves the drought tolerance of poplar. Gene 2014, 545, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hanin, M.; Brini, F.; Ebel, C.; Toda, Y.; Takeda, S.; Masmoudi, K. Plant dehydrins and stress tolerance: Versatile proteins for complex mechanisms. Plant Signal. Behav. 2011, 6, 1503–1509. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, X.; Zhang, L. Structural and functional dynamics of dehydrins: A plant protector protein under abiotic stress. Int. J. Mol. Sci. 2018, 19, 3420. [Google Scholar] [CrossRef] [Green Version]
- Meyers, B.C.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R.W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef] [Green Version]
- Meyers, B.C.; Morgante, M.; Michelmore, R.W. TIR-X and TIR-NBS proteins: Two new families related to disease resistance TIR-NBS-LRR proteins encoded in Arabidopsis and other plant genomes. Plant J. 2002, 32, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Nandety, R.S.; Caplan, J.L.; Cavanaugh, K.; Perroud, B.; Wroblewski, T.; Michelmore, R.W.; Meyers, B.C. The role of TIR-NBS and TIR-X proteins in plant basal defense responses. Plant Physiol. 2013, 162, 1459–1472. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-M.; Chen, M.; Sun, L.; Wang, Y.; Yin, J.; Liu, J.; Sun, X.-Q.; Hang, Y.-Y. Genome-wide identification and evolutionary analysis of NBS-LRR genes from dioscorea rotundata. Front. Genet. 2020, 11, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, F.; Zhu, S.; Li, X. The maize NBS-LRR gene ZmNBS25 enhances disease resistance in rice and Arabidopsis. Front. Plant Sci. 2018, 9, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Takahashi, N.; Kanno, Y.; Suzuki, H.; Aoi, Y.; Takeda-Kamiya, N.; Toyooka, K.; Kasahara, H.; Hayashi, K.; Umeda, M.; et al. The Arabidopsis NRT1/PTR FAMILY protein NPF7.3/NRT1.5 is an indole-3-butyric acid transporter involved in root gravitropism. Proc. Natl. Acad. Sci. USA 2020, 117, 31500–31509. [Google Scholar] [CrossRef]
- Liu, X.; Bush, D.R. Expression and transcriptional regulation of amino acid transporters in plants. Amino Acids 2006, 30, 113–120. [Google Scholar] [CrossRef]
- Schwacke, R.; Grallath, S.; Breitkreuz, K.E.; Stransky, E.; Stransky, H.; Frommer, W.B.; Rentsch, D. LeProT1, a transporter for proline, glycine betaine, and gamma-amino butyric acid in tomato pollen. Plant Cell 1999, 11, 377–392. [Google Scholar]
- Kohl, S.; Hollmann, J.; Blattner, F.R.; Radchuk, V.; Andersch, F.; Steuernagel, B.; Schmutzer, T.; Scholz, U.; Krupinska, K.; Weber, H.; et al. A putative role for amino acid permeases in sink-source communication of barley tissues uncovered by RNA-seq. BMC Plant Biol. 2012, 12, 154–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Hasan, M.; Li, Y.; Liao, C.; Zheng, H.; Liu, R.; Li, X. Asymmetric transcriptomic signatures between the cob and florets in the maize ear under optimal- and low-nitrogen conditions at silking, and functional characterization of amino acid transporters ZmAAP4 and ZmVAAT3. J. Exp. Bot. 2015, 66, 6149–6166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.M.; Ma, H.L.; Yu, L.; Wang, X.; Zhao, J. Genome-wide survey and expression analysis of amino acid transporter gene family in rice (Oryza sativa L.). PLoS ONE 2012, 7, e49210. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Torre, A.R.; Sekhwal, M.K.; Neale, D.B. Selective Sweeps and Polygenic Adaptation Drive Local Adaptation along Moisture and Temperature Gradients in Natural Populations of Coast Redwood and Giant Sequoia. Genes 2021, 12, 1826. https://doi.org/10.3390/genes12111826
De La Torre AR, Sekhwal MK, Neale DB. Selective Sweeps and Polygenic Adaptation Drive Local Adaptation along Moisture and Temperature Gradients in Natural Populations of Coast Redwood and Giant Sequoia. Genes. 2021; 12(11):1826. https://doi.org/10.3390/genes12111826
Chicago/Turabian StyleDe La Torre, Amanda R., Manoj K. Sekhwal, and David B. Neale. 2021. "Selective Sweeps and Polygenic Adaptation Drive Local Adaptation along Moisture and Temperature Gradients in Natural Populations of Coast Redwood and Giant Sequoia" Genes 12, no. 11: 1826. https://doi.org/10.3390/genes12111826