
Abstract
Multiple sclerosis (MS) is a heterogenous autoimmune disease in which autoreactive lymphocytes attack the myelin sheath of the central nervous system. B lymphocytes in the cerebrospinal fluid (CSF) of patients with MS contribute to inflammation and secrete oligoclonal immunoglobulins1,2. Epstein–Barr virus (EBV) infection has been epidemiologically linked to MS, but its pathological role remains unclear3. Here we demonstrate high-affinity molecular mimicry between the EBV transcription factor EBV nuclear antigen 1 (EBNA1) and the central nervous system protein glial cell adhesion molecule (GlialCAM) and provide structural and in vivo functional evidence for its relevance. A cross-reactive CSF-derived antibody was initially identified by single-cell sequencing of the paired-chain B cell repertoire of MS blood and CSF, followed by protein microarray-based testing of recombinantly expressed CSF-derived antibodies against MS-associated viruses. Sequence analysis, affinity measurements and the crystal structure of the EBNA1–peptide epitope in complex with the autoreactive Fab fragment enabled tracking of the development of the naive EBNA1-restricted antibody to a mature EBNA1–GlialCAM cross-reactive antibody. Molecular mimicry is facilitated by a post-translational modification of GlialCAM. EBNA1 immunization exacerbates disease in a mouse model of MS, and anti-EBNA1 and anti-GlialCAM antibodies are prevalent in patients with MS. Our results provide a mechanistic link for the association between MS and EBV and could guide the development of new MS therapies.
Similar content being viewed by others


Main
The presence of oligoclonal bands (OCBs) in CSF and the efficacy of therapies that deplete B cells emphasize the importance of B cells in the pathobiology of MS2. Antiviral antibodies against mumps, measles, varicella-zoster virus (VZV) and EBV are often present in MS4,5, but their relevance is unclear. Anti-EBNA1 antibody titres can be detected in nearly 100% of patients with MS before the development of clinical symptoms, which providesevidence for an epidemiological link between MS and EBV6. Symptomatic infectious mononucleosis during EBV infection increases the risk for developing MS7. Molecular mimicry between virus and self-antigens is a potential mechanism that might explain this association8. Antibodies against certain EBNA1 regions have been found in patients with MS, including the region covering residues 365–426 (refs. 5,9,10,11,12), which we describe here in our identification of molecular mimicry between EBNA1 and GlialCAM. The potential significance of this mimicry in the pathophysiology of MS is described in detail.
The B cell repertoire in MS CSF
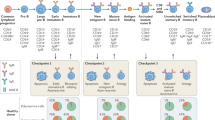
CSF and blood samples were obtained from patients with MS during the onset of disease (clinically isolated syndrome, n = 5) or an acute episode of relapsing–remitting MS (n = 4). Patients with a CSF pleocytosis of >10 cells µl–1 were selected (Extended Data Table 1, Supplementary Discussion). Single B cells were sorted by flow cytometry (Extended Data Fig. 1a, b). and characteristic phenotypic differences of B cells in blood and CSF were observed13,14. These included high plasmablast (PB) counts in CSF compared to blood (Extended Data Fig. 1c, d), different expression levels of α4 integrin and HLA-DR in PBs but not in non-PB B cells (Extended Data Fig. 1e–j, Supplementary Table 1) and high abundance of immunoglobulin G (IgG) in CSF PBs (Fig. 1a, Extended Data Fig. 1k, l).

a–c, Single-cell BCR repertoire sequencing data. a, Individual repertoires from blood PBs (top row) and CSF B cells (second row) of n = 9 patients with MS, and CSF B cells of n = 3 control patients with other neuroimmunological diseases (bottom row). Numbers indicate the number of sequences. For the inner circle, coloured wedges represent clonal expansions and the grey area represents singleton antibody sequences. The outer circle represents immunoglobulin classes, and sequence locations in the outer circle correspond to the inner circle. b, Clonality analysis. The percentage of clonal sequences in CSF B cells are shown, comparing BCR repertoires of control individuals (n = 3) to patients with MS (n = 9). Mean ± s.d. values of each group are shown. **P = 0.0091, two-tailed Mann–Whitney test. c, IGHV gene distribution in blood versus CSF PBs. Each dot represents the use of one IGHV gene across n = 9 MS repertoires in the respective compartments. Linear regression line and 95% confidence interval are shown. IGHV1-2, ***P = 5.6 × 10−4; IGHV4-59, ***P = 9.2 × 10−4; IGHV3-7, *P = 0.025; unpaired two-tailed Student’s t-tests, Holm–Sidak corrected for multiple comparisons. d, Mass spectrometry data of purified CSF immunoglobulins showing variable chain sequences that were uniquely identified by mass spectrometry in the singleton BCR sequences versus clonal sequences (peptide-spectral matches cut-off ≥ 1), mean ± s.d. of n = 9 patients with MS. ***P = 0.0002, two-tailed Mann–Whitney test.
We sorted PBs from blood and B cells from matched CSF samples by flow cytometry and sequenced their full-length paired heavy-chain (HC) and light-chain (LC) VDJ regions15. A total of 13,578 paired sequences from blood PBs and 1,689 from CSF B cells passed filter thresholds. The CSF repertoire was highly clonal (Fig. 1a, Extended Data Figs. 2a, 3), which suggests that antigen-specific proliferation of select clones occurs within the CSF. Notably, CSF repertoires in three individuals without MS but with neuroinflammatory conditions (Extended Data Table 1) did not exhibit such extensive clonality and high IgG use (Fig. 1a, b, Extended Data Fig. 2b–d). Although the amount of somatic hypermutation (SHM) in immunoglobulin HC-V (IGHV) and LC-V (IGLV) genes did not differ between PBs in blood and CSF (Extended Data Fig. 2e), the lengths of HC-complementarity determining region 3 (CDR3) were longer in CSF PBs than in blood. This suggests that there is ongoing intrathecal SHM in the CSF (Extended Data Fig. 2f). Consistent with previous reports16,17,18,19, repertoires in the CSF were skewed towards the use of five IGHV genes (Fig. 1c, Extended Data Fig. 2g–i), which indicates that select autoantigens drive PB survival and proliferation in CSF.
CSF PBs produce oligoclonal IgG
Immunoglobulin levels are increased in the CSF of patients with MS, and the presence of a few highly abundant OCBs is a hallmark of the disease. To determine whether clonal PBs are the source of intrathecal immunoglobulins, we isolated immunoglobulins from CSF, characterized them by mass spectrometry and compared the spectra with their corresponding antibody repertoire sequence datasets. For 87% of the clonal sequences, peptides matching variable-chain sequences unique to the patient were identified, whereas this occurred for only 40% of singleton sequences (Fig. 1d). Highly abundant immunoglobulins, defined by the detection of ≥10 peptide-spectral matches, aligned almost exclusively to clonally expanded B cells (Extended Data Fig. 2j) and to PBs, which display more clonality than non-PB B cells (Extended Data Fig. 2k, l). This result indicates that clonally expanded PBs are the predominant source of CSF OCBs.
CSF B cell-encoded antibodies bind EBV
A total of 148 B cell receptor (BCR) sequences from MS CSF, each representative of a clonal expansion, were expressed as monoclonal antibodies (mAbs) (Extended Data Fig. 3). MS CSF mAbs showed low polyreactivity (polyreactive mAbs from patients with MS were 5.4% compared with 15.4% for controls) (Extended Data Figs. 4, 5b). To test for antiviral reactivities, mAbs were probed on protein microarrays containing 2 EBV viral lysates, 23 latent and lytic EBV proteins, 240 peptides spanning four prominent EBV proteins, and lysates of 7 other MS-associated viruses, including measles, rubella and VZV4 (Fig. 2a, b, Extended Data Figs. 5a, 6, 7a, Supplementary Tables 2, 3). One-third of the expressed mAbs bound to EBV proteins and peptides and about 20% to other viruses, in particular to VZV and cytomegalovirus (Fig. 2a). Notably, half of the VZV-reactive antibodies cross-reacted to cytomegalovirus and EBV, which indicates that there are conserved epitopes among herpesviruses.

a, Protein microarray showing MS CSF mAb reactivities (z-scores) to viral lysates and EBV proteins. b, MS CSF mAb reactivities against EBNA1 peptides. mAbs with highest reactivities are shown (n = 36 (a) and n = 37 (b) out of n = 148). Data from one experiment, eight technical replicates. E, early; IE, immediate-early; L, late lytic stage. Red represents mAb MS39p2w174 and antigen EBNA1/MS-associated region. c, Western blot of recombinant EBNA1 (full-length and truncated proteins) stained on separate blots with commercial anti-C-terminal EBNA1 antibody or MS39p2w174. d, ELISA-based overlapping peptide scan of MS39p2w174 binding to EBNA1 peptides (20 mers, 13 amino acid overlap). Mean ± s.d. of n = 4 independent experiments, each measured in duplicate. e, ELISA-based alanine scan of EBNA1AA386–405, logo representation shows the contribution of each residue to the binding of MS39p2w174. f–j, Crystal structures of MS39p2w174-Fab in complex with EBNA1AA386–405. f, Cartoon representation showing EBNA1AA389–401 in the binding groove. Peptide in red, HC in green/blue, and LC in purple, yellow and orange; the CDR loop colours correspond to annotations in g. g, View of the binding groove from the top. Surface representation of the Fab with EBNA1AA389–401 in stick representation. h, Cartoon and stick representation outlining close interactions. Major hydrogen-bond-forming residues are represented as sticks. Hydrogen bonds < 3.2 Å are represented as black dashed lines. i, Magnification of the peptide in the hydrophobic cage. j, Magnification of the region around Arg396 to emphasize polar contacts of HC residues with Arg396 and Arg397. k, l, Bio-layer interferometry measurement of the affinity of MS39p2w174 and germline to full-length EBNA1 protein. k, Mean Kd ± s.d. values of one representative experiment of three independent experiments. **P = 0.0043, unpaired two-tailed Student’s t-test. l, Association and dissociation curves corresponding to k.
We also found that mAbs from six out of nine patients with MS bound to the EBV transcription factor EBNA1 (Fig. 2a, Extended Data Fig. 5a), and mAbs that bound EBNA1 peptides were identified in eight out of nine patient samples (Fig. 2b, Extended Data Fig. 6). Anti-EBNA1 reactivity has been implicated in MS epidemiology20, and the amino acid region AA365–425 is the target of stronger antibody responses in patients with MS than in non-affected individuals5,9,10,11,12. Protein and peptide microarrays revealed that the mAb MS39p2w174 bound EBNA1 within this MS-associated region at AA386–405 (Fig. 2b, Extended Data Fig. 6). This epitope was confirmed by western blot analysis using full-length and truncated EBNA1 proteins (Fig. 2c) and by ELISA-based EBNA1 peptide scans (Fig. 2d). Alanine scanning identified the Pro/Arg-rich region AA394–399 as the central epitope for MS39p2w174 (Fig. 2e, Extended Data Fig. 7b).
Structure of the EBNA1–CSF Fab complex
Although the presence of antibodies against the broader EBNA1 region AA365–425 has been described in MS5,9,10,11,12, their relevance remains elusive. We solved the crystal structure of EBNA1AA386–405 in complex with the Fab region of MS39p2w174 at a resolution of 2.5 Å (Protein Data Bank (PDB) identifier 7K7R)21 (Fig. 2f–j, Extended Data Fig. 7c–e). Close interactions of the EBNA1 residues P394–P398 were observed with all CDRs except for LC-CDR2. Residues Tyr31 and Tyr38 on LC-CDR1 together with Trp38 on HC-CDR1 and with Pro108, Pro109 and Tyr114 on HC–CDR3 create a hydrophobic cage for the two N-terminal proline residues Pro394 and Pro395 and the proximal side chain of Arg396 of the peptide (Fig. 2h, i). The C-terminal end of the antibody-binding groove is wider, and Pro398 is carried by a large aromatic tryptophan residue (Trp114 in HC-CDR1) (Fig. 2g, h). The central arginine residues Arg395 and Arg396 form hydrogen bonds with residues on HC-CDR2, HC-CDR3 and HC framework region 2.
The IGHV encoding gene of MS39p2w174 is IGHV3-7, one of the IGHV chains over-represented in CSF (Fig. 1c). Of note, all but two of the residues that directly interact with EBNA1 are unmutated germline residues (Extended Data Fig. 7f, g). We hypothesized that the unmutated ancestor of MS39p2w174 (germline) might have an innate propensity to bind EBNA1AA386–405. Indeed, germline was significantly more polyreactive than MS39p2w174 (Extended Data Fig. 4), and bio-layer interferometry revealed that germline binds EBNA1 with almost the same affinity as MS39p2w174 (dissociation constant (Kd) values of 1.99 ± 0.63 nM (mean ± s.d.) for MS39p2w174 compared with 4.19 ± 0.76 nM for germline) (Fig. 2k, l), which indicates that SHM is not required for effective EBNA1 binding and that naive B cells have EBNA1 specificity.
Molecular mimicry of EBNA1 and GlialCAM
We probed the mAb MS39p2w174 on HuProt microarrays, which contained over 16,000 proteins spanning more than 80% of the human proteome22, and discovered that MS39p2w174 binds GlialCAM (Fig. 3a). GlialCAM is an immunoglobulin superfamily cell adhesion molecule expressed in the central nervous system (CNS) by astrocytes and oligodendrocytes23,24,25,26. Earlier proteomics studies of MS brain lesions by our laboratory revealed that GlialCAM is expressed in chronic-active plaques27. MS39p2w174 was also probed using a human proteome-wide phage display immunoprecipitation and sequencing (PhIP–seq) library28. MS39p2w174 did not demonstrate high enrichment for any single peptide, which suggests that it has low affinity for multiple native peptides (Extended Data Table 2). Peptide motif analysis identified a common Pro/Arg-rich motif that closely resembles the central epitope in EBNA1 (AA395–399; Fig. 2e and Extended Data Fig. 8a). Comparing the PhIP–seq results with the HuProt results yielded two overlapping targets: the ubiquitously expressed actin filament associated protein 1 (AFAP1, which does not have the Pro/Arg-rich motif) and the CNS protein GlialCAM (encoded by HEPACAM). Binding of MS39p2w174 to the intracellular domain (ICD; AA262–416) of GlialCAM was confirmed by ELISA (Fig. 3b) and western blotting (Fig. 3c). MS39p2w174 showed distinct glial staining of mouse brain, including radial Bergmann glia in the cerebellum, the glia limitans, and perivascular glial cells in the hippocampus and the brainstem (Fig. 3d, and Extended Data Fig. 8b). MS39p2w174 stained rat oligodendrocytes and transgenic GlialCAM-expressing K562 cells, in which GlialCAM generated tight junctions (Extended Data Fig. 8c–e).

a, Protein microarray showing the ratio of MS39p2w174/mean control reactivity (left, 89–911) and raw mean fluorescent intensities (right, 1–36,450). b, ELISA of MS39p2w174 to EBNA1AA328–641, and GlialCAM full-length and ICD. Mean ± s.d. of n = 3 independent experiments, each in triplicate. c, Western blots of GlialCAM full-length, ICD and ECD stained with anti-GlialCAM and MS39p2w174. d, Immunofluorescence images of mouse brains stained with MS39p2w174. Top, cerebellum. Middle, inferior colliculus (ic) and occipital cortex (occ) with perivascular glia (open arrowheads) and glia limitans (filled arrowheads) indicated. Bottom, dentate gyrus with perivascular staining (open arrowheads). Scale bar, 40 µm (bottom), 60 µm (middle) or 150 µm (top). e, f, Bio-layer interferometry of MS39p2w174 and germline with EBNA1. e, Mean Kd ± s.d. values of a representative of three independent experiments. *P = 0.012, unpaired two-tailed Student’s t-test, f, Association and dissociation curves corresponding to e. g, Alignment of EBNA1AA386–405 and GlialCAMAA370–389. h, i, Disorder prediction (using predictor of natural disordered regions (PONDR)) for EBNA1 (h) and GlialCAM (i). j, ELISA of MS39p2w174 to EBNA1AA386–405 and GlialCAMAA370–389, non-phosphorylated versus phosphorylated. Means are from duplicates of n = 2 independent experiments, representative of 5 experiments. k, l, Bio-layer interferometry of MS39p2w174 with GlialCAM peptides. k, Mean Kd ± s.d. values of one representative of three independent experiments. ****P < 0.0001, unpaired two-tailed analysis of variance, Tukey corrected. l, Association and dissociation curves corresponding to k. m, Protein microarray of mAb reactivities (mean fluorescence intensities) to GlialCAM and EBV proteins and peptides from one experiment (eight replicates). n–p, ELISAs of human plasma reactivities against EBNA1AA328–641 (n; ****P < 0.0001), EBNA1AA386–405 (o; **P < 0.0044) and GlialCAM protein (p; ***P < 0.0002). Unpaired two-tailed Mann–Whitney test, mean ± s.d. from n = 20 unaffected individuals and n = 36 patients with MS. q, ELISA of plasma reactivity against EBNA1AA386–405, blocked with scrambled peptide, EBNA1AA386–405 and GlialCAMAA370–389 pSer376. Mean optical density (OD) fold-change over unblocked ± s.d. of n = 9 patients with MS, in quadruplicate. **P = 0.0037; ****P < 0.0001, Kruskal–Wallis test, Dunn-corrected for multiple comparisons.
Although we demonstrated that MS39p2w174 and its unmutated germline ancestor bound EBNA1 with similar affinity (Fig. 2k, l), their binding affinities to GlialCAM differed significantly. The affinity of MS39p2w174 to GlialCAM was increased by ≥10-fold (Kd of 1.99 ± 0.63 nM for EBNA1 compared with 190 ± 17 pM for GlialCAM). By contrast, germline bound GlialCAM with lower affinity (Kd of 4.19 ± 0.76 nM for EBNA1 compared with 10.46 ± 4.12 nM for GlialCAM) (Fig. 3e, f). Evidently, although germline harbours a propensity to bind to EBNA1, SHM of MS39p2w174 increased its affinity to the CNS mimic GlialCAM by an order of magnitude.
GlialCAM phosphorylation enables mimicry
The EBNA1 epitope AA386–405 is located between the long N-terminal Gly/Ala-rich low-complexity region (AA90–380) of the protein and its highly structured DNA-binding domain (AA461–607; PDB identifier 1B3T (ref. 29)). The GlialCAM region AA337–385, identified by phage display (Extended Data Table 2), is located at the C-terminal end of the ICD and contains a Pro-rich region that resembles the central epitope of EBNA1 (Fig. 3g). Intracellular B cell autoantigens have been described in several autoimmune diseases30,31,32. MS39p2w174 reacted with both proteins on western blots under denaturing conditions (Figs. 2c, 3c), which suggests that both antibody-binding regions are linear epitopes. This result is consistent with predictions that both epitopes are located in intrinsically disordered regions of their respective proteins (Fig. 3h, i).
Nevertheless, although MS39p2w174 bound with similar affinity to both the EBNA1 protein and the EBNA1AA386–405 peptide (Kd of 1.99 ± 0.63 nM for the protein and 2.67 ± 0.078 nM for the peptide), its binding affinity for the native peptide GlialCAMAA370–389 was three orders of magnitude lower than for GlialCAM protein (Kd of 190 ± 17 pM for the protein and 302 ± 31 nM for the peptide). Part of this discrepancy could be due to multimerization of GlialCAM33. Moreover, the ICD of GlialCAM is heavily phosphorylated33,34,35 (Extended Data Fig. 8f and PhosophSite36; https://www.phosphosite.org), and post-translational modifications (PTMs) often determine antibody–antigen interactions37. We determined whether phosphorylation at one of the four serine residues surrounding the central epitope region (Ser376, Ser377, Ser383 and Ser384) could increase the binding affinity of MS39p2w174 to GlialCAMAA370–389. Indeed, phosphorylation at Ser376 (pSer376) increased MS39p2w174 binding by about 50-fold (Kd of 302 ± 0.078 nM for the native peptide and 6.1 ± 0.27 nM for the pSer376 peptide), and additional phosphorylation of Ser377 further enhanced the binding affinity (Kd of 3.73 ± 0.15 nM for the pSer376/pSer377 peptide) (Fig. 3j–l). By contrast, citrullination of arginine residues Arg373, Arg380 and Arg387 did not alter binding to GlialCAMAA370–389 (Extended Data Fig. 8g). The important residue Arg397 in EBNA1AA386–405, which engages in two hydrogen bonds with Glu64 at HC-CDR2 (Fig. 2h, j), is replaced with alanine in GlialCAMAA370–389 (Ala381) (Fig. 3g). This probably explains the differential binding affinity to EBNA1 and GlialCAM peptides. Phosphorylation at Ser376 probably promotes binding by providing new polar interactions to the proximal LC, possibly with Arg36, a positively charged residue that is mutated from asparagine in germline (Fig. 2h, Extended Data Fig. 7g). Together, our results show that post-translational phosphorylation enables cross-reactivity of anti-EBNA1 MS39p2w174 to GlialCAM.
Anti-GlialCAM IgG reactivity in MS
To determine whether the observed anti-GlialCAM reactivity of MS39p2w174 represents a broader phenomenon in MS, we tested the remaining 147 clonal MS mAbs for reactivity against GlialCAM protein and peptides spanning GlialCAMAA315–395. Ten additional mAbs from seven patients bound the ICD and seven more from four patients bound the extracellular domain (ECD) (Fig. 3m), which demonstrates that MS PBs generate antibodies against multiple GlialCAM epitopes. We then tested plasma samples from patients with MS (n = 36) and from non-affected individuals (n = 20) for reactivity to EBNA1 and GlialCAM. As expected, all samples from patients with MS and the majority samples from healthy individuals exhibited plasma reactivity to EBNA1 protein (Fig. 3n). Reactivity to EBNA1AA386–405 and to GlialCAM was significantly increased in patients with MS (Fig. 3o, p). Increased plasma IgG levels against EBNA1AA386–405, GlialCAMAA370-–389 and phosphorylated GlialCAMAA370–389 pSer376 were confirmed in a second independent patient cohort (MS, n = 71, controls, n = 50) (Extended Data Fig. 9a). Similarly, we detected increased plasma IgG levels against GlialCAM, GlialCAMAA370–389 and phosphorylated GlialCAMAA370-389 pSer376 in a third cohort (MS, n = 67, controls, n = 31) (Extended Data Fig. 9b). We next addressed whether reactivity against EBNA1AA386–405 could be blocked by GlialCAM. Inhibition of anti-EBNA1AA386–405 reactivity by GlialCAMAA370–389 pSer376 in nine high-reactivity samples was observed (Fig. 3q, Extended Data Fig. 9c), which indicates that this molecular mimicry is prevalent in a subset of patients with MS.
EBNA1 immunization aggravates EAE
To assess the effect of an anti-EBNA1AA386–405 immune response on neuroinflammation, we used mice with experimental autoimmune encephalomyelitis (EAE), the mouse model of MS. SJL/J mice were immunized with scrambled control peptide or EBNA1AA386–405. Three weeks later, EAE was induced by a second immunization of the respective peptides mixed with proteolipid protein (PLPAA139–151). Mice in the EBNA1AA386–405 group generated robust antibody responses against both EBNA1AA386–405 (Fig. 4a) and GlialCAM ICD (Fig. 4b). The antibody response to PLPAA139–151 was unaltered in both groups (Extended Data Fig. 10a). In addition, EBNA1AA386–405 immunization induced a strong CD4+ T cell response, whereas the T cell response against PLPAA139–151 remained stable in both groups (Extended Data Fig. 10b). EBNA1AA386–405 stimulated the secretion of B cell stimulatory T helper 1 cytokines, including interferon-γ (IFNγ), tumour necrosis factor and interleukin-12 (IL-12), as well as IL-6 and IL-10 (Extended Data Fig. 10c–g), but suppressed the key T helper 17 cytokine IL-17 (Extended Data Fig. 10h). Clinically, the EBNA1-treated group exhibited more severe paresis (Fig. 4c), with enhanced CNS immune cell infiltration (Fig. 4d, e) and demyelination (Extended Data Fig. 10i, j).

a, b, ELISA data showing mouse plasma IgG responses at the indicated time points before and after EAE induction for mice immunized with scrambled peptide (n = 10 mice) or EBNA1AA386–405 (n = 10 mice). Plasma reactivities are shown against EBNA1AA386–405 (a; mean OD450 nm fold change ± s.d., ****P < 0.0001, unpaired two-tailed Mann–Whitney test, Holm–Sidak corrected for multiple comparisons) and GlialCAM ICD (b; mean OD450 nm fold change ± s.d., **P = 0.0022, *P = 0.0152, unpaired two-tailed Mann–Whitney test, Holm–Sidak corrected for multiple comparisons), representative of three independent experiments, each carried out as triplicate measurements. c, EAE scores of mice immunized with scrambled peptide (n = 9) or EBNA1AA386-405 (n = 7), 3 weeks before and on the day of EAE immunization (day 0). Means of clinical scores ± s.e.m., *P < 0.05 unpaired two-tailed Mann–Whitney test. d, e, Spinal cord histology. d, Representative haematoxylin and eosin (H&E)-stained spinal cords from mice immunized with scrambled peptide (top) or EBNA1AA386-405 (bottom). Scale bars, 50 μm (right) or 200 μm (left). e, Statistical evaluation of H&E scores. Means of at least four coronal spinal cord sections per mouse and mean ± s.d. for each group (n = 9). **P = 0.0012, unpaired two-tailed Mann–Whitney test. f, Flow cytometry data of PBMCs from healthy individuals (n = 6, blue) and patients with MS (n = 7, red) showing the per cent of IFNγ+granzyme B+CD8+ T cells in all CD8+ T cells. Mean ± s.e.m. values are shown for the respective groups. Significance levels were assessed by two-way analysis of variance, followed by false discovery rate calculation using the two-stage step-up method of Benjamini, Krieger and Yekutieli, *Significant at false discovery rate of < 0.1.
To further assess T cell reactivity in humans, peripheral blood mononuclear cells (PBMCs) from patients with MS (n = 7) with increased anti-EBNA1 and anti-GlialCAM titres were stimulated with EBNA1 and GlialCAM proteins and peptides and compared with PBMCs from healthy individuals. The CD4+ T cells trended towards more IFNγ expression after stimulation with EBNA1AA386–405 and GlialCAM ECD and ICD proteins, which resembled the CD4+ T cells in mice (Extended Data Fig. 10k, l). In CD8+ T cells, high expression of IFNγ and granzyme B indicated a robust CD8+ T cell response against EBNA1 in both groups, whereas only CD8+ T cells from patients with MS responded to GlialCAM ICD and GlialCAM ECD (Fig. 4f). One patient (MS16) (Fig. 3o, p) showed extraordinarily high counts of IFNγ+granzyme B+CD8+ T cells after stimulation with EBNA1, GlialCAM ICD and GlialCAMAA370–389 (Extended Data Fig. 10m).
Together, these results show that anti-GlialCAM antibodies are generated in response to EBNA1AA386–405 immunization. Furthermore, immunization of mice with both EBNA1AA386–405 and PLPAA139–151 enhanced CNS immune cell infiltration and demyelination, two prominent features of human MS pathology. In addition to anti-GlialCAM antibody titres, our human T cell data suggest an important role for CD8+ T cells in the immune response against GlialCAM.
Discussion
Viral triggers of MS and other autoimmune diseases have long been the subject of intense investigation, but evidence for their functional relevance is scarce8. By utilizing paired-chain BCR repertoire sequencing, rational selection of clonal antibody sequences and three independent high-throughput proteomics platforms, we identified a mAb from CSF of a patient with MS that binds the MS-associated epitope EBNA1AA386–405 with high affinity and cross-reacts with GlialCAM. We demonstrated the presence of cross-reactive EBV EBNA1 and GlialCAM antibodies in 20–25% of patients with MS and showed that immunization of EAE mice with this EBNA1 epitope exacerbated autoimmune demyelination. Our findings demonstrate a mechanistic link between EBV infection and the pathobiology of MS.
Our analysis of the single-cell paired-chain BCR repertoire from blood and CSF of patients with MS revealed features of intrathecal oligoclonal expansion with ongoing SHM that were substantially different from CSF BCR repertoires in other neuroinflammatory diseases. PBs in the CSF of patients with MS are highly clonal, with long CDR3 regions and skew towards the use of five IGHV genes16,17,18,19,38,39. IGHV3-7 has been previously observed in MS CSF and OCBs17,19,39,40,41. The discovery that the IGHV3-7-based mAb MS39p2w174 cross-binds EBV EBNA1 and GlialCAM provides a direct link between IGHV3-7 and MS pathobiology.
Structural and binding data of MS39p2w174 from the CSF of a patient with MS and its unmutated germline ancestor showed that only germline bound EBNA1AA486–405 with high affinity, and that SHM is required for high-affinity cross-reactivity with GlialCAM. Owing to the CNS-restricted expression of GlialCAM, it is likely that immature IGHV3-7-expressing B cells encoding a germline precursor of MS39p2w174 entered the CNS and CSF space, encountered GlialCAM antigen and then underwent affinity maturation that generated clones encoding high-affinity anti-GlialCAM antibodies. This is concordant with our BCR repertoire analysis, which revealed a highly clonal and skewed repertoire in CSF with low polyreactivity that is indicative of a select group of B cells undergoing SHM, probably driven by perpetual activation by CNS GlialCAM.
The crystallographic structure revealed that Arg397 in EBNA1AA386–405 engages in two hydrogen bonds with HC-CDR2 and is probably responsible for a notable part of the high binding affinity between MS39p2w174 and EBNA1. We showed that phosphorylation of GlialCAM at Ser376 restores high affinity. Epitopes altered by PTMs have been described as targets for autoimmune responses, including citrullinated proteins in rheumatoid arthritis and phosphorylated Ro/La in systemic lupus erythematosus37,42. Tissue-specific differences of PTMs could contribute to a lack of central tolerance. Several kinases have been described as risk genes for MS, including MERTK, MAPK1, MAPK3 and TYK2, which potentially contribute to alternative phosphorylation patterns in the CNS43.
In addition, we showed that immunization of EAE mice with EBNA1AA386–405 generates a robust B cell response against GlialCAM and aggravates EAE. Our finding of activated PBs in the CSF that express exceptionally high levels of HLA-DR suggest that these B cells present antigens and exchange inflammatory signals with T follicular helper cells.
In summary, we presented a detailed picture of the B cell repertoire in MS CSF and blood and demonstrated that activated intrathecal IgG+ PBs undergo affinity maturation. Analysis of a CSF-derived mAb that targets EBNA1AA386–405 revealed molecular mimicry to GlialCAM. These findings provide a mechanistic link between EBV infection and the pathobiology of MS.
Methods
Study design and human participants
Patient samples were collected at Stanford University and the University of Heidelberg. Relapsing–remitting MS was diagnosed according to the current McDonald criteria44,45. None of the patients met the diagnostic criteria for neuromyelitis optica spectrum disorder, in particular spinal lesions spanning ≥3 segments46. Patients were tested for antibodies against aquaporin-4 and myelin oligodendrocyte glycoprotein and showed negative results. All included patients had increased CSF white blood cell counts (≥10 cells µl–1), and blood-contaminated CSF samples were excluded by visual and microscopy inspection. Paired peripheral blood and CSF samples were obtained at the time of clinical onset (clinically isolated syndrome) or during an acute relapse. All but one patient had not received any MS-specific treatment before sample collection (Extended Data Table 1). As this study does not explore group differences, no specific randomization strategy has been applied for the selection of study participants. MS is a disease that predominantly affects women, which is reflected in our study cohort which contained paired CSF and blood samples from n = 8 female and n = 1 male MS patients. The study size is in line with prior repertoire studies investigating both, single-cell and bulk B cell repertoires in MS and other diseases13,16. All experimental protocols were approved by the institutional review board (IRB) of Stanford University (IRB number 34529) and the ethics committee of the medical faculty of the University of Heidelberg (IRB number S-466/2015). Written informed consent was obtained from each patient.
Cell preparation, antibody staining and flow cytometry cell sorting
CSF was centrifuged immediately after lumbar puncture and cells were counted. PBMCs were isolated from heparinized blood by density gradient centrifugation using Ficoll Plus medium (Cytiva). Cells were magnetically separated with anti-CD19 magnetic beads (Dynabeads CD19 Pan B cell isolation kit, Invitrogen), then stained according to standard protocols using antibodies against the following cell surface markers: CD20 (clone L27, dilution 1:10), CD38 (clone HB7, dilution 1:30) and IgD (clone IA6-2, dilution 1:20) from BD Biosciences); CD3 (clone OKT3, dilution 1:60), CD19 (clone HIB19, dilution 1:20), CD27 (clone O323, dilution 1:20), IgM (clone MHM-88, dilution 1:40), HLA-DR (clone L243, dilution 1:100) and α4 integrin (clone 9F10, dilution 1:100) from BioLegend; IgA (clone IS11-8E10, dilution 1:20) from Miltenyi Biotec; and Sytox blue (dilution 1:500) from ThermoFisher Scientific. Single cells were sorted using a FACSAria II cell sorter (BD Biosciences) with FACSDiva (v.8.0, BD Biosciences) into 96-well PCR plates (Bio-Rad). For single-cell repertoire sequencing, PBs were sorted from PBMCs (plasmablast gate (4.12%) in panel 5 in the representative flow cytometry plots shown in Extended Data Fig. 1a). All B cells were sorted from CSF (B cell gate (73.9%) in panel 4 in the representative flow cytometry plots shown in Extended Data Fig. 1b). FlowJo v.10.7.1 (BD) and R v.3.6.1 were used to evaluate flow cytometry data.
Single-cell BCR repertoire sequencing
BCR repertoire sequencing was carried out using our in-house developed plate-bound single-cell sequencing technology as previously described15,47,48. In brief, reverse transcription with oligo-dT was carried out in separate wells, attaching unique well-ID barcodes by template switching activity of Maxima Reverse Transcriptase (ThermoFisher Scientific). Barcoded cDNA from each plate was pooled and amplified in three consecutive PCRs, including attaching plate-specific barcodes and sequencing adapters. PCRs were carried out separately for the HC of IgG, IgA and IgM, as well as for the κ-chain for λ-LC, and separate libraries were generated from each. Samples were then gel-purified, cleaned with Ampure XP beads (Beckman Coulter) and sequenced on an Illumina MiSeq (Illumina) with 2 × 330 paired-end reads.
Sequence analysis
The MiSeq FASTQ workflow was used for Fastq generation and plate demultiplexing. R v.3.6.1 was used for custom analyses. Paired reads of sequences that passed quality thresholds were stitched and separated by plate and well IDs. Similar reads sharing the same plate and well IDs were clustered into operational taxonomic units49. Consensus sequences were aligned to germline variable-chain immunoglobulin sequences with IMGT HighV-QUEST (v.1.3.1)50, which reports V, D and J germline genes, HC and LC CDR3 lengths, and non-silent mutation counts and locations. Clonal expansions were defined on the basis of sharing the same HC and LC V and J genes and exhibiting >70% amino acid identity within the HC and LC CDR3s. Per cent clonality represents the per cent of all sequences that fulfil these criteria. To calculate IGHV, IGLV, IGHJ and IGLJ gene usage, the per cent abundance of each particular gene was calculated in blood and CSF PBs of each patient, and means were calculated across patients. Genes that were present in fewer than three CSF samples were excluded from this analysis. Although our sequencing method preferentially captures PB sequences owing to higher amounts of immunoglobulin mRNA (Extended Data Fig. 2m), enough non-PB B cell sequences passed filter thresholds to compare the non-PB repertoire to the PB repertoire in seven patients (Extended Data Fig. 2a, b). For patient samples MS12 and C6, only PBs were captured (while gating on all B cells), and for MS39, only PBs were sorted. For phylogenetic analysis, sequences were binned according to their HC V-gene family and V gene. Concatenated LC and HC were then aligned with Muscle51 and clustered with PhyML52 using maximum-likelihood clustering. Each tree-partition was rooted by their HC V-gene. Phylogenetic trees were drawn in Python using the ETE 3 toolkit53.
Peptide identification by mass spectrometry
Immunoglobulins were purified from 1.5 ml of CSF samples with Protein A (ThermoFisher Scientific). The purified IgG samples were reduced with 0.02 M dithiothreitol at 57 °C for 1 h, alkylated with 0.05 M iodoacetamide at room temperature (RT) in the dark, and digested with trypsin overnight at RT. Peptides were extracted and desalted as previously described54. An aliquot of the peptide mixtures was loaded onto an Acclaim PepMap 100 precolumn (75 μm × 2 cm, C18, 3 μm, 100 Å) in-line with an EASY-Spray, PepMap column (75 μm × 50 cm, C18, 2 μm, 100 Å) with a 5-μm emitter using the autosampler of an EASY-nLC 1000 (ThermoFisher Scientific). The peptides were gradient eluted into a Lumos Fusion Tribrid (ThermoFisher Scientific) mass spectrometer using a 120-min gradient from 5% to 35% solvent B (90% acetonitrile, 0.5% acetic acid), followed by 10 min from 35% to 45% solvent B and 10 min from 45% to 100%. High-resolution full mass spectrometry spectra were acquired with a resolution of 120,000, an AGC target of 4 × 105, a maximum ion time of 50 ms and a scan range of 400–1,800 m/z. Following each full mass spectrometry scan, as many data-dependent HCD tandem mass spectrometry spectra were acquired in the orbitrap as possible in a 3-s cycle time. Monoisotopic precursor selection (MIPS) was set to peptide, precursors with a charge state of 2–7 and minimum intensity of 5 × 104 were selected for tandem mass spectrometry. Dynamic exclusion was set to 60 s after a single selection. All tandem mass spectrometry spectra were collected using the following instrument parameters: resolution of 30,000, an AGC target of 105, maximum ion time of 120 ms, 2 microscans, 1.6 m/z isolation window and normalized collision energy of 32.
The tandem mass spectrometry spectra were searched against the respective peptide-specific database, including common contaminant proteins, using the search engine Byonic54. The search parameters were set to trypsin allowing two missed cleavages, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine and deamidation on glutamine and asparagine. Peptides mapping to variable regions of IgG were manually verified. To include only sequence-specific peptides, peptides that aligned to non-immunoglobulin or constant-region sequences were excluded from the analysis, as were peptides that aligned to the repertoire of multiple patient samples. Included were peptides that aligned to one variable sequence in a single patient. Peptides that aligned to more than one variable sequence in a single patient were included if all matching sequences were exact matches or clonally related, in which case the peptide was counted as representative for all matches. Counts of identical or non-identical peptide-spectral matches per sequence were tallied for each sequence. Sequences that had >1 or >10 matching peptides were presented as a percentage of all sequences (Fig. 1d and Extended Data Fig. 2j, k). The mass spectrometry files are accessible at MassIVE (https://massive.ucsd.edu) under accession number MSV000086842.
Selection and recombinant expression of mAbs
Representative antibodies from the largest clonal B cell expansions in the CSF of each patient were selected for recombinant expression. In patients with more than 10 large clonal expansions, sequences were preferentially chosen based on their usage of one of the 11 most abundant IGHV genes in the CSF (Extended Data Fig. 3). HC and LC variable sequences were custom-generated (IDT) and cloned into pFuse vectors (Invivogen) that contained human IgG constant region or kappa or lambda constant regions, respectively. Fab HC was expressed in in-house plasmids that contained the HC constant-region C1 up to Cys103. Plasmids were transfected into Expi293T cells using Expifectamine (ThermoFisher Scientific). Authenticity of 293T cell lines were certified by the supplier. Cell cultures were tested regularly for mycoplasma contamination. Culture medium was collected after 4 days and 7 days after transfection. mAbs and Fabs were purified with protein A and protein G resins, respectively (ThermoFisher Scientific). Antibody concentrations were measured with a nanodrop spectrophotometer (ThermoFisher Scientific) and hIgG quantitation ELISAs (Bethyl Laboratories) and checked for purity on SDS protein gels with Coomassie staining.
Protein expression and purification
The following EBNA1 proteins and peptides were obtained: full-length AA1–641 (Abcam), AA328–641 (Virion Serion) and AA408–641 (ProspecBio). The following GlialCAM proteins and peptides were obtained: full-length AA34–416 (OriGene) and ECD AA34–234 (Novoprotein). GlialCAM ICD AA262–416 was expressed in Escherichia coli with an N-terminal His-Tag. In brief, the codon-optimized sequence was cloned into a pet30(+) vector, expressed in BL21 chemically competent E. coli (Sigma Aldrich) to an optical density of 600 nm, and induced with IPTG (Sigma Aldrich) for 3 h at 37 °C. Cell pellets were disrupted by sonication, and proteins were purified with complete His-Tag purification resins (Roche Life Science), followed by size-exclusion purification (Cytiva). For all other used peptides and proteins see Supplementary Tables 2–4.
Planar protein microarrays
Protein microarrays were generated as previously described55,56 (https://web.stanford.edu/group/antigenarrays/). In brief, peptides, recombinant proteins and lysates were diluted at the indicated concentrations in a 1:1 solution of PBS/water and protein printing buffer (ArrayIt) (Supplementary Tables 2–4), aliquoted into 384-well plates and printed on SuperEpoxy slides using a NanoPrint LM210 system (ArrayIt). Two independent quadruplicates of each analyte were spotted, and some proteins were used in several versions/preparations from different sources (Supplementary Table 2). Ready-made HuProt Arrays v.3.1 were obtained from CDI Labs. Arrays were circumscribed with a hydrophobic marker, blocked overnight at 4 °C in PBS containing 3% FCS and 0.1% Tween-20, and incubated with individual mAbs at a concentration of 1 µg ml–1 for 1 h at 4 °C, then washed twice for 20 min each in blocking buffer on a rotating shaker. Arrays were then incubated with Cy-3-conjugated secondary goat anti-human IgG (0.8 μg ml–1; Jackson ImmunoResearch) for 1 h at 4 °C, then washed twice for 30 min each in blocking buffer, twice for 30 min each in PBS, and twice for 15 s each in water. Arrays were spun dry and scanned with a GenePix 4000B scanner (Molecular Devices). Median pixel intensities for each fluorescent spot were determined with GenePix Pro-3.0 software (Molecular Devices). Z-scores for each row of antigens were calculated for viral antigens, whereas raw intensities were analysed for GlialCAM arrays. Heatmaps were generated with Morpheus software (The Broad Institute; https://software.broadinstitute.org/morpheus).
PhIP–seq
PhIP–seq was performed using a human proteome-wide library, expressing overlapping 49-amino-acid peptides with a 24-amino-acid sliding window approach starting at the N terminus. In brief, 2 μl (1 μg ml–1) of substrate antibody was diluted 1:100 in blocking buffer for two sequential rounds of immunoprecipitation. After the second round of immunoprecipitation and amplification in E. coli, next-generation sequencing libraries were prepared for paired-end 150 base next-generation DNA sequencing on the Illumina Hi-Seq platform as previously described28,57. After alignment of the reads to the reference peptide sequences, quality control was performed, and only reads present at an abundance of fewer than ten reads per hundred thousand were carried forward. The number of reads mapping to each peptide were then counted and individually scored as a percentage of the total.
ELISA
The following cytokine ELISA kits were used according to the manufacturers’ instructions: mouse IL-6, IL-10, IL-12, IFNγ and TNF (BD Biosciences), and IL-17A (ThermoFisher Scientific). For protein and peptide ELISAs, MaxiSorp 384-well plates (ThermoFisher Scientific) were coated with 1 µg ml–1 peptide or protein in carbonate-bicarbonate buffer at 4 °C overnight, then washed 6 times with PBST (PBS + 0.05 % Tween20), blocked with blocking buffer (PBS + 1% BSA) for 1 h, and mAbs were applied at 1 µg ml–1 in blocking buffer. Human and mouse plasma samples were diluted 1:100 and T cell supernatants 1:4 in blocking buffer. After overnight incubation at 4 °C, plates were washed again 6 times with PBST and then secondary antibody HRP-conjugated goat anti-human IgG (Jackson ImmunoResearch) was applied for 1 h at RT. After six additional washes with PBST, plates were developed with TMB substrate (ThermoFisher Scientific), stopped with 1 N sulfuric acid and read on a SpectraMax Paradigm Microplate Reader (Molecular Devices). For plasma ELISAs with blocking of plasma IgG, MaxiSorp 384-well plates were coated with 2 µg ml–1 recombinant Protein G (Acro Biosystems) at 4 °C overnight, then washed six times with PBST and incubated with 1:100 diluted plasma at 4 °C overnight. Plates were again washed 6 times with PBST, then incubated with the respective blocking peptides at 10 µg ml–1 for 2 h at room temperature. Biotinylated EBNA1AA385–405 was added at 1 µg ml–1 and incubated for 1 h at room temperature. Plates were washed again six times with PBST, incubated with HRP-conjugated streptavidin (BioLegend) for 1 h at room temperature and developed with TMB substrate as described above.
Western blotting
Western blots were run according to standard protocols. In brief, purified proteins were boiled in Laemmli buffer with 10% β-mercaptoethanol for 5 min, run on 4–12% Criterion XT Bis-Tris protein Gels (Bio-Rad) and then transferred onto a nitrocellulose membrane using a Trans-Blot Turbo semi-dry transfer system (Bio-Rad). The blots were stained with MS39p2w174 at 10 µg ml–1 or with mouse anti-EBNA1 antibody (Biorbyt) or mouse anti-GlialCAM antibody (R&D Systems) followed by secondary HRP-conjugated goat anti-human IgG and anti-mouse IgG (Jackson ImmunoResearch). The western blots in Figs. 2c and 3c were performed on separate membranes. Coomassie gels were run concomitantly, fixed with 10% methanol and 7% acetic acid, and stained according to standard protocols. Uncropped western blot and Coomassie images are available in Supplementary Fig. 1.
Fluorescent immunohistochemistry on mouse brain slices and immunofluorescence on primary cultured rat oligodendrocytes
An adult mouse (F1 generation of FVB × C57BL/6 cross) was transcardially perfused with 4% paraformaldehyde (PFA) and post-fixed in 4% PFA overnight at 4 °C. After sucrose equilibration, the brain was blocked in OCT and sectioned at 12 µm on a standard cryostat. Sections were permeabilized and blocked in PBS containing 10% lamb serum and 0.1% Triton X-100. Sections were immunostained with concomitantly expressed control mAb anti-DSG3 (Acc: HQ338093.1 and HQ338094.1; 18 µg ml–1), MS39p2w174 (18 µg ml–1) or PBS in blocking buffer overnight at 4 °C. Sections were washed five times with PBS over 1 h and counterstained with anti-human IgG for 1 h at room temperature (2 µg ml–1 Alexa Fluor 488, Jackson ImmunoResearch). Nuclei were stained with DAPI at 1:2,000 for 5 min at room temperature. Rat oligodendrocyte precursor cells were prepared from rat embryos followed by panning and in vitro differentiation into primary rat oligodendrocytes58. Cells on coverslips were permeabilized with ice-cold 100% methanol for 10 min, blocked with 10% donkey serum for 1 h at room temperature and then stained with isotype control (anti-DSG3) or MS39p2w174 at 10 µg ml–1 in 1% donkey serum for 1 h at RT, before incubation with secondary Alexa Fluor 647 donkey anti-human IgG antibody (Jackson ImmunoResearch) for 1 h at RT. Confocal images were taken with a Zeiss LSM 880 confocal microscope using Zen software (Zeiss).
Bio-layer interferometry
Association and dissociation constants of mAbs to proteins and peptides were measured using bio-layer interferometry on an Octet QK device (Fortebio/Sartorius) according to standard protocols. For peptide kinetics, biotinylated peptides were bound to high-precision streptavidin (SAX) biosensors (peptide concentration in solution of 100 nM), and the mAbs MS39p2w174 and germline were probed as analytes in concentrations ranging from 10 to 270 nM. For protein kinetics, mAbs were bound to anti-hIgG Fc capture (AHC) biosensors (mAb concentration in solution of 20 nM), and proteins were probed as analytes in concentrations ranging from 1.56 to 125 nM. Data were analysed with bio-layer interferometry analysis software (Fortebio/Sartorius, v.7.1) and GraphPad prism (v.8.4). Buffer controls were subtracted, and curves were fitted globally for each group consisting of all concentrations of the same ligand. Kd values ± s.d. as well as association–dissociation curves are reported and plotted with GraphPad prism (v.8.4). Kd, Kon and Koff values are shown in Supplementary Table 6. Kd values reported in Figs. 2k and 3e, k are mean ± s.d. values from at least three serial dilutions from one representative out of three independent experiments.
Prediction of protein disorder
Order and disorder along the amino acid sequences of EBNA1 and GlialCAM were analysed using predictor of natural disordered regions (PONDR; WSU Research Foundation)59 with the VSL2 algorithm.
Crystallization of antibody–antigen complexes
EBNA1AA386–405 20mer peptides (>98% purity) (Sigma Aldrich) were mixed with MS39p2w174-Fab (15 mg ml–1) in a 7.5:1 molar ratio and incubated overnight. Crystals for MS39p2w174-Fab and EBNA1AA386–405 grew in 0.48 M sodium citrate, 0.72M sodium/potassium phosphate and 3% MPD (v/v) in 0.1 M HEPES, pH 6.9 (Extended Data Fig. 7c). Crystals were collected, cryo-protected with a quick dip in a cryo-solution containing the well solution with 25% glycerol and flash-cooled in liquid nitrogen. Data were collected at beamline SSRL 12-2 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, and processed and scaled using XDS/aimless and Staraniso60,61. Crystals belonged to space group I222 (a = 119.66 Å, b = 137.56 Å, c = 179.00 Å, α = β = γ = 90°) and contained two Fab–peptide complexes per asymmetric unit (Extended Data Fig. 7d). Phaser was used for molecular replacement62 with the model structure 4LRI (PDB)63 stripped of all CDR loops. Loops were reconstructed with Coot64 and structures were refined with phenix.refine65,66 in iterations with Coot. Measurements and figure design were performed using Pymol (v.2.1)67. The structure was deposited in PDB (https://www.rcsb.org)21 with the PDB identifier 7K7R.
Mouse immunization, EAE and histology
All animal experiments were performed in accordance with state and federal guidelines and regulations, and approved by the Stanford Institutional Animal Care and use Committee. Eight-week-old female SJL/J and FVB × C57BL/6 mice were purchased from The Jackson Laboratory. The mice were housed in recyclable individually ventilated cages, with a 12-h light/dark cycle, at a temperature of 21 °C and with 50% humidity. Mice were immunized subcutaneously with 200 µg per mouse of EBNA1AA386–405 (peptide sequence: SQSSSSGSPPRRPPPGRRPF) or scrambled control peptide (peptide sequence: SPSRPGRSRSRGSPFPQPSP) (10 mice per group), mixed with 100 µg per mouse of CpG (ODN1826, Invivogen) in 100 µl per mouse incomplete Freund’s adjuvant (BD Difco). Three weeks later, EAE was induced by subcutaneous immunization with 200 µg per mouse of PLPAA139–151, mixed with the same peptides used in the first immunization, in 100 µl of incomplete Freund’s adjuvant, supplemented with 200 µg per mouse of mycobacterium tuberculosis (strain H37 RA, BD Difco). Serum samples were obtained by retro-orbital blood draws 3 days before the first and second immunizations (day –24 and day –3), and at the termination of the experiment (day 50). Mice were weighed every day, and disease severity was assessed in a blinded manner according to a 5-point standard scoring system: 0, no clinical signs; 1, loss of tail tone; 2, hindlimb weakness; 3, complete hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, moribund or dead. Mice were euthanized on day 50 after induction of EAE by deep anaesthesia with intraperitoneal injections of 0.01 ml g–1 body weight 7.2% xylazine (Bayer Healthcare) and 10.8% ketamine (Pfizer). Lymph nodes and spleens were removed, and mice were then perfused with 10 ml PBS and 20 ml 4% PFA (Electron Microscopy Sciences). Brains and spinal cords were removed, stored in 4% PFA overnight followed by 30% sucrose in PBS. Tissues were embedded in paraffin, sectioned and stained for H&E as well as Luxol fast blue according to standard protocols. Infiltration of inflammatory cells into the spinal cord on H&E slides was assessed by a blinded investigator by counting lesions of infiltrating cells per slide, taking lesion size into account. The following scoring system was used: 1, small infiltrate (<10 cells); 2, medium infiltrate (<100 cells); 3, large infiltrate (>100 cells). Demyelination was assessed by a blinded investigator according to a histological score68 as follows: 0.5, single demyelinated spot; 1, several spots; 2, confluent sites of demyelination; 3, extensive demyelination, less than half of a spinal cord; 4, demyelination of more than half of the spinal cord; and 5, extensive demyelination affecting >85% of the total white matter of the spinal cord.
In vitro human T cell stimulation with proteins and peptides
Cryopreserved PBMCs were thawed and stabilized overnight at 37 °C. The cells were pre-incubated for 30 min at 37 °C, 5% CO2 with polymyxin B (Sigma Aldrich) at a concentration of 10 μg ml–1. The cells were then incubated for 16 h with 100 μM of each recombinant protein or peptide in the presence of 2 μg ml–1 of anti-human CD28 (clone CD28.2, BD Biosciences) and anti-CD49d (clone 9F10, BioLegend) antibodies and IL-2 (50 IU ml–1, Peprotech) and IL-7 (5 ng ml–1, Peprotech). To detect intracellular staining, eBioscience Protein Transport Inhibitor cocktail (500X, ThermoFisher Scientific) was added during the final 5 h of culture. After 16 h, the cells were labelled with Fixable Viability Stain 510 (BD Biosciences) for live cell staining and fluorophore conjugated anti-CD3 (clone SK7, BD Biosciences), CD4 (clone RPA-T4, BD Biosciences), CD8 (clone RPA-T8, BD Biosciences), granzyme B (clone GB11, BD Biosciences), IFNγ (clone B27, BD Biosciences) and IL-17A (clone BL168, BD Biosciences) antibodies and detected using a BD LSR Fortessa.
Data analysis and statistics
The publicly available dataset from Han et al.27 was searched for abundance of GlialCAM in MS lesions. The old NCBI Protein Database accession number Q8N7I3 was found, which was annotated in 2008 as unknown hypothetical protein and has since been replaced by accession number Q14CZ8. GlialCAM was identified with 2.5 mean spectral counts (MSCs) in control tissue, 1.3 MSCs in chronic plaques, 1.8 MSCs in acute plaques and 8 MSCs in chronic-active plaques.
GraphPad Prism v.8.4.1 and R v.3.6.1 were used for statistical analyses. Statistical tests used are indicated in the respective section of the Methods or in the figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The genomic datasets analysed during the study have been uploaded to the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with accession number PRJNA780931. Mass spectrometry data are available at MassIVE (https://massive.ucsd.edu) with accession number MSV000086842. Structural data are available at PDB (https://www.rcsb.org) with the identifier 7K7R. Source data are provided with this paper.
References
Cencioni, M. T., Mattoscio, M., Magliozzi, R., Bar-Or, A. & Muraro, P. A. B cells in multiple sclerosis—from targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 17, 399–414 (2021).
Hauser, S. L. et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 376, 221–234 (2017).
Bar-Or, A. et al. Epstein–Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol. Med. 26, 296–310 (2020).
Jarius, S. et al. The MRZ reaction as a highly specific marker of multiple sclerosis: re-evaluation and structured review of the literature. J. Neurol. 264, 453–466 (2017).
Wang, Z. et al. Antibodies from multiple sclerosis brain identified Epstein–Barr virus nuclear antigen 1 & 2 epitopes which are recognized by oligoclonal bands. J. Neuroimmune Pharmacol. 16, 567–580 (2021).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Nielsen, T. R. et al. Effects of infectious mononucleosis and HLA-DRB1*15 in multiple sclerosis. Mult. Scler. 15, 431–436 (2009).
Tengvall, K. et al. Molecular mimicry between anoctamin 2 and Epstein–Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl Acad. Sci. USA. 116, 16955–16960 (2019).
Ruprecht, K. et al. Multiple sclerosis: the elevated antibody response to Epstein–Barr virus primarily targets, but is not confined to, the glycine–alanine repeat of Epstein–Barr nuclear antigen-1. J. Neuroimmunol. 272, 56–61 (2014).
Jafari, N. et al. No evidence for intrathecal IgG synthesis to Epstein–Barr virus nuclear antigen-1 in multiple sclerosis. J. Clin. Virol. 49, 26–31 (2010).
Salzer, J. et al. Epstein–Barr virus antibodies and vitamin D in prospective multiple sclerosis biobank samples. Mult. Scler. 19, 1587–1591 (2013).
Sundqvist, E. et al. Epstein–Barr virus and multiple sclerosis: interaction with HLA. Genes Immun. 13, 14–20 (2012).
Ramesh, A. et al. A pathogenic and clonally expanded B cell transcriptome in active multiple sclerosis. Proc. Natl Acad. Sci. USA 117, 22932–22943 (2020).
Kowarik, M. C. et al. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J. Neurol. 261, 130–143 (2014).
Tan, Y.-C. et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 66, 2706–2715 (2014).
Palanichamy, A. et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 6, 248ra106 (2014).
von Büdingen, H.-C. et al. B cell exchange across the blood–brain barrier in multiple sclerosis. J. Clin. Invest. 122, 4533–4543 (2012).
Rounds, W. H. et al. MSPrecise: a molecular diagnostic test for multiple sclerosis using next generation sequencing. Gene 572, 191–197 (2015).
Bankoti, J. et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann. Neurol. 75, 266–276 (2014).
Mescheriakova, J. Y., van Nierop, G. P., van der Eijk, A. A., Kreft, K. L. & Hintzen, R. Q. EBNA-1 titer gradient in families with multiple sclerosis indicates a genetic contribution. Neurol. Neuroimmunol. Neuroinflamm. 7, e872 (2020).
Berman, H. M. et al. The Protein Data Bank. Acta Crystallogr. D 58, 899–907 (2002).
Jeong, J. S. et al. Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics 11, O111.016253 (2012).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Gilbert, A., Vidal, X. E., Estevez, R., Cohen-Salmon, M. & Boulay, A.-C. Postnatal development of the astrocyte perivascular MLC1/GlialCAM complex defines a temporal window for the gliovascular unit maturation. Brain Struct. Funct. 224, 1267–1278 (2019).
Favre-Kontula, L. et al. GlialCAM, an immunoglobulin-like cell adhesion molecule is expressed in glial cells of the central nervous system. Glia 56, 633–645 (2008).
López-Hernández, T. et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88, 422–432 (2011).
Han, M. H. et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081 (2008).
O’Donovan, B. et al. High-resolution epitope mapping of anti-Hu and anti-Yo autoimmunity by programmable phage display. Brain Commun 2, fcaa059 (2020).
Bochkarev, A., Bochkareva, E., Frappier, L. & Edwards, A. M. The 2.2 Å structure of a permanganate-sensitive DNA site bound by the Epstein–Barr virus origin binding protein, EBNA1. J. Mol. Biol. 284, 1273–1278 (1998).
Casciola-Rosen, L. A., Anhalt, G. & Rosen, A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 179, 1317–1330 (1994).
Pisetsky, D. S. & Lipsky, P. E. New insights into the role of antinuclear antibodies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 16, 565–579 (2020).
Schellekens, G. A. et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 43, 155–163 (2000).
Moh, M. C., Zhang, C., Luo, C., Lee, L. H. & Shen, S. Structural and functional analyses of a novel Ig-like cell adhesion molecule, hepaCAM, in the human breast carcinoma MCF7 cells. J. Biol. Chem. 280, 27366–27374 (2005).
Britton, D. et al. Quantification of pancreatic cancer proteome and phosphorylome: indicates molecular events likely contributing to cancer and activity of drug targets. PLoS ONE 9, e90948 (2014).
Herskowitz, J. H. et al. Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J. Proteome Res. 9, 6368–6379 (2010).
Hornbeck, P. V., Chabra, I., Kornhauser, J. M., Skrzypek, E. & Zhang, B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4, 1551–1561 (2004).
Zavala-Cerna, M. G. et al. The clinical significance of posttranslational modification of autoantigens. Clin. Rev. Allergy Immunol. 47, 73–90 (2014).
Bashford-Rogers, R. J. M., Smith, K. G. C. & Thomas, D. C. Antibody repertoire analysis in polygenic autoimmune diseases. Immunology 155, 3–17 (2018).
Obermeier, B. et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 14, 688–693 (2008).
Singh, V. et al. Cerebrospinal-fluid-derived immunoglobulin G of different multiple sclerosis patients shares mutated sequences in complementarity determining regions. Mol. Cell. Proteomics 12, 3924–3934 (2013).
Colombo, M. et al. Accumulation of clonally related B lymphocytes in the cerebrospinal fluid of multiple sclerosis patients. J. Immunol. 164, 2782–2789 (2000).
Terzoglou, A. G., Routsias, J. G., Avrameas, S., Moutsopoulos, H. M. & Tzioufas, A. G. Preferential recognition of the phosphorylated major linear B-cell epitope of La/SSB 349–368 aa by anti-La/SSB autoantibodies from patients with systemic autoimmune diseases. Clin. Exp. Immunol. 144, 432–439 (2006).
International Multiple Sclerosis Genetics Consortium & The Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Thompson, A. J. et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 17, 162–173 (2018).
Polman, C. H. et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69, 292–302 (2011).
Wingerchuk, D. M. et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85, 177–189 (2015).
Tan, Y.-C. et al. High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin. Immunol. 151, 55–65 (2014).
Blum, L. K. et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 48, 874–884 (2018).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Alamyar, E., Duroux, P., Lefranc, M.-P. & Giudicelli, V. IMGT® tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 882, 569–604 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Huerta-Cepas, J., Serra, F. & Bork, P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016).
Bern, M., Kil, Y. J. & Becker, C. Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics Chapter 13, Unit13.20 (2012).
Robinson, W. H. et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 8, 295–301 (2002).
Kuerten, S. Autoantibodies against central nervous system antigens in a subset of B cell-dominant multiple sclerosis patients. Proc. Natl Acad. Sci. USA 117, 21512–21518 (2020).
Schubert, R. D. et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 25, 1748–1752 (2019).
Emery, B. & Dugas, J. C. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868 (2013).
Obradovic, Z. et al. Predicting intrinsic disorder from amino acid sequence. Proteins 53, 566–572 (2003).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 (2013).
Tickle, I. STARANISO: use of a WebGL-based 3D interactive graphical display to represent and visualise data quality metrics for anisotropic macromolecular diffraction data. Acta Crystallogr. A 75, e162 (2019).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Fouts, A. E. et al. Mechanism for neutralizing activity by the anti-CMV gH/gL monoclonal antibody MSL-109. Proc. Natl Acad. Sci. USA 111, 8209–8214 (2014).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012).
The PyMOL Molecular Graphics System, Version 2.1, Schrödinger, LLC.
Lanz, T. V. et al. Tryptophan-2,3-dioxygenase (TDO) deficiency is associated with subclinical neuroprotection in a mouse model of multiple sclerosis. Sci Rep. 7, 41271 (2017).
Acknowledgements
We thank W. Wick for support with sample collection; B. Bell for advice on crystallization; G. Harauz for insightful discussions on poly-proline motifs; and the staff at SSRL for assistance with data collection. This work was supported by NIH R01 AR063676 and U19 AI110491 to W.H.R., the Juvenile Diabetes Research Foundation and Lupus Research Alliance Funding to W.H.R. and T.V.L., the German Research Foundation (DFG, LA3657/1) to T.V.L., Atara to L.S. and P.P.H., and the German Research Foundation to M.P. (DFG, project 406052676; PL-315/5-1). The mass spectrometry experiments were in part supported by the NYU Grossman School of Medicine and a shared instrumentation grant (NIH 1S10OD010582-01A1). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract number DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by NIH GIGMS P30GM133894. C.M.B. is a Hanna H. Gray Fellow at the Howard Hughes Medical Institute. The contents of this publication are solely the responsibility of the authors and do not represent the official views of the NIGMS or the NIH.
Author information
Authors and Affiliations
Contributions
Conceptualization: T.V.L., L.S. and W.H.R. Methodology: T.V.L., R.C.B., P.P.H., K.M.J., D.F., R.A.F., A.M.G., R.D.S., B.T., V.C.C., B.M.U., J.-S.M., M.I. and J.B.Z. Software: T.V.L., R.C.B., D.F., K.M.J. and V.C.C. Validation: T.V.L., R.C.B., K.M.J., B.M.U., R.J.M.B.-R., K.C.G., L.S. and W.H.R. Formal analysis: T.V.L., R.C.B., K.M.J., D.F., A.M.G., C.M.B., V.C.C. and B.M.U. Investigation: T.V.L., R.C.B., P.P.H., D.F., G.-S.N., C.M.B., R.D.S., I.A.H., S.E.V., B.T., V.C.C., J.-S.M. and M.I. Resources: T.V.L., P.P.H., D.F., B.T., J.E.D., C.B.L., L.B.K., B.M.U., M.R.W., M.S.A., J.L.D., M.P., K.C.G., L.S. and W.H.R. Data curation: T.V.L., R.C.B., K.M.J., D.F., V.C.C. and B.M.U. Writing (original draft): T.V.L. Writing (review and editing): T.V.L., R.C.B., P.P.H., L.S. and W.H.R. Visualization: T.V.L., R.C.B. and K.M.J. Supervision: T.V.L. and W.H.R. Project administration: T.V.L. and W.H.R. Funding acquisition, T.V.L., P.P.H., B.T.A., M.P., L.S. and W.H.R.
Corresponding author
Ethics declarations
Competing interests
W.H.R. owns equity in, serves as a consultant to and is a member of the Board of Directors of Atreca, Inc. L.S. owns equity in and serves as a consultant to Atreca, Inc. Stanford University is in the process of applying for a patent, US Patent and Trademark Office Serial No. 63/131,581, covering anti-EBV antibodies generated by sequencing B cell repertoires, which lists T.V.L. and W.H.R. as inventors. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature thanks Paul Farrell, George Georgiou, Hartmut Wekerle and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Analysis of B cell phenotypes in MS blood and CSF.
a–l, Flow cytometry data, a,b, representative flow cytometry plots are shown for a, blood and b, CSF. c, Plasmablasts as percent of all B cells in MS blood and CSF, means ± SD of n = 9 patient samples, **P = 0.004, two-tailed Mann-Whitney test. d, Non-plasmablast B cell subsets as percent of all B cells in blood (red) and CSF (blue), means ± SD of n = 8 patient samples, ***P = 0.0006, two-tailed Mann-Whitney test, Holm-Sidak corrected for multiple comparisons. e, Integrin alpha-4 expression in non-plasmablast B cells (red) and plasmablasts (blue), mean MFI ± SD of n = 9 patient samples, ****P < 0.0001, **P = 0.0013, two-way ANOVA, Tukey adjusted for multiple comparisons, f, representative histogram showing integrin alpha-4 expression in non-plasmablast B cells (red) and plasmablasts (blue) in blood (top panel) and CSF (lower panel), g, HLA-DR expression in non-plasmablast B cells (red) and plasmablasts (blue) in blood and CSF, mean MFI ± SD of n = 9 patient samples, ****P < 0.0001, ***P = 0.0002, two-way ANOVA, Tukey adjusted for multiple comparisons, and h, representative histogram showing HLA-DR expression in non-plasmablast B cells (red) and plasmablasts (blue) in blood (top panel), and CSF (lower panel). i, HLA-DR expression in patients carrying HLA-DRB1*15:01 (HLA-DR15, n = 5) vs. other HLA-genotypes (non-HLA-DR15, n = 4) in i, blood, and j, CSF, mean MFI ± SD, significance levels calculated with two-way ANOVA, k,l, Immunoglobulin classes in k, non-plasmablast B cells and l, plasmablasts in blood (red) and CSF (blue), mean MFI ± SD of n = 9 patient samples, ****P < 0.0001, two-way ANOVA, Holm-Sidak adjusted for multiple comparisons. Plasmablasts, PB; unswitched memory B cells, UM; switched memory B cells, SM; double negative B cells, DN.
Extended Data Fig. 2 Extended BCR repertoire data.
a–i, Single-cell BCR repertoire sequencing data, a, individual repertoires from all CSF B cells (top row) and subdivided into CSF plasmablasts (middle row) and non-plasmablast B cells (bottom row) of n = 9 MS patients. b, Individual repertoires of all CSF B cells (top row) and subdivided into CSF plasmablasts (middle row) and non-plasmablast B cells (bottom row) of n = 3 control patients. Numbers indicate number of sequences, inner circle: colored wedges represent clonal expansions and grey area represents singleton antibody sequences, outer circle: immunoglobulin classes, red: IgG, blue: IgA, green: IgM, sequence locations in outer circle correspond to inner circle. No non-plasmablast B cells were sorted for MS12 and C5. Only plasmablasts were sorted for MS39. c, Clonality, percent of clonal sequences in CSF B cells are shown, comparing BCR repertoires of control patients (n = 3) to MS patients (n = 9). Data corresponds to data shown in (Fig. 1b) and is separated into immunoglobulin classes IgG (left), IgA (center), and IgM (right). Means ± SD of individuals’ repertoires are shown. d, Immunoglobulin class distribution, percent of IgG (left), IgA (center), and IgM (right) of all CSF B cells are shown for n = 3 control patients and n = 9 MS patients. Means ± SD of individuals’ repertoires are shown. e, IGHV and IGLV cumulated mutation count in plasmablasts in blood (red) vs. CSF (blue), means ± SD of n = 9 patients samples. f, Mean HC CDR3 lengths (amino acid sequences) of plasmablasts in blood (red) vs. CSF (blue), means ± SD of n = 9 patient samples. g–i, Immunoglobulin gene distribution in blood vs. CSF plasmablasts for g, IGLV, IGKV1-33, ****P < 10−6, IGLV3-21, ****P = 3 x 10−6 according to unpaired two-tailed Student’s t tests, Holm-Sidak adjusted for multiple comparisons, h, IGHJ, and i, IGLJ. Each dot represents the usage of one gene across n = 9 MS patient repertoires in the respective compartments. Linear regression lines and 95% confidence intervals are shown. j, Mass spectrometry data of purified CSF immunoglobulins, showing variable chain sequences that could be uniquely identified in singleton BCR sequences vs. plasmablast sequences, peptide-spectral matches (PSM) cutoff ≥10, means ± SD of n = 9 MS patients, **P = 0.0012. k, l, Same mass spectrometry data set as in (j), showing variable chain sequences that could be uniquely identified in non-plasmablast BCR sequences vs. plasmablast sequences, means ± SD of n = 7 MS patients, k, PSM cutoff ≥1, **P = 0.007, l, PSM cutoff ≥10, *P = 0.037. m, Single-cell sequencing efficacy in non-plasmablast B cells (red) vs. plasmablasts (blue) in CSF. Fraction of sequences that passed filter thresholds are shown as percentages of the number of sorted cells in the respective group, means ± SD of n = 8 patient samples (no non-PB value for MS39). c, d, j–l, P according to unpaired two-tailed Mann-Whitney test. e–i, P according to unpaired two-sided Student’s t-test. Immunoglobulin heavy V gene, IGHV; Immunoglobulin heavy J gene, IGHJ; Immunoglobulin light V gene, IGLV; Immunoglobulin light J gene, IGLJ; peptide-spectral matches, PSM.
Extended Data Fig. 3 Phylogenetic trees of B cells from MS blood and CSF.
Blood plasmablasts (top rows) and CSF B cells (bottom rows) of n = 9 MS patients and CSF B cells of n = 3 control patients are shown. Each node represents the full-length heavy chain and light chain sequence of a single B cell. Trees are binned according to their IGHV families and genes, then the concatenated heavy chain and light chain sequences are clustered. IgG (red), IgA (blue), IgM (green). Smaller brighter circles indicate singleton B cells, larger darker circles indicate clonal expansions. Arrows indicate sequences that were expressed as mAbs, numbers indicate V-gene mutation loads in heavy and light chains. Immunoglobulin heavy V gene, IGHV.
Extended Data Fig. 4 Polyreactivity of recombinantly expressed antibodies.
a, ELISA data showing reactivity of recombinant mAbs against LPS (top), human insulin (middle), and dsDNA (bottom). Reactivity is represented in the order of decreasing reactivity to LPS in MS mAbs and control mAbs, respectively. Measurements were carried out in duplicates at 0.1, 1, and 10 µg/ml mAb concentrations and the area under the curve (AUC) for each mAb is shown from one experiment. Commercial anti-LPS antibody (cyan), MS39p2w174 (red), germline (orange), control mAbs (blue).
Extended Data Fig. 5 MS CSF mAb reactivity to EBV and GlialCAM antigens.
a, mAb reactivities to EBV virus lysates and recombinant EBV proteins as well as to other virus lysates. Z-scores for each antigen are shown, measurement of one microarray experiment, measured in 8 technical replicates. b, mAb reactivities to LPS, Insulin, and dsDNA to assess polyreactivity. Z-scores of area under the curve (AUC) of ELISA measurements at antibody concentrations of 0.1, 1, and 10 µg/ml are shown, each measurement was carried out in duplicates. c, mAb reactivities to GlialCAM proteins, peptides, and phosphorylated or citrullinated peptides. Mean reactivities (mean fluorescence intensity counts) are shown from one microarray experiment, measured in 8 technical replicates. Immediate early latency stage protein, IE; early, E; late, L; intracellular domain, ICD; extracellular domain, ECD; phosphorylated Serine, pSer; citrulline residue, Cit; _B - _E: duplicate probes of same / similar lysates and proteins (different preparations or batches).
Extended Data Fig. 6 MS CSF mAb reactivity to EBV peptides.
a, mAb reactivities to EBV peptides. Z-scores for each antigen are shown, measurement of one microarray experiment, measured in 8 technical replicates. Intracellular domain, ICD; extracellular domain, ECD; peptide mix, PM.
Extended Data Fig. 7 mAb reactivity to EBV peptides and extended structural data for the EBNA1AA386-405 / MS39p2w174-Fab complex.
a, mAb reactivities of selected reactive mAbs against the selected reactive peptide antigens. Z-scores for each antigen are shown, measurement of one microarray experiment, measured in 8 technical replicates. b, ELISA-based alanine-scan on EBNA1AA386-405, corresponding to (Fig. 2e). Mean OD (450 nm) ± SD from three independent experiments, each carried out in triplicates. c, 20x image of protein crystals in hanging drop. d, Asymmetric unit containing two peptide-Fab complexes in a diagonal orientation, heavy chain (red/pink), light chain (blue/cyan), peptide (black/gray). e, EBNA1AA386-405 peptide and its 2mFoDFc map (contoured at 1σ) are shown, depicted on heavy chain (cyan) and light chain (pink) in surface representation. f, g, Amino acid sequences of variable regions of f, mAb MS39p2w174 heavy chain and g, light chain. Bold font: CDR, regular font framework regions. Of the germline variable genes (bottom rows), only residues that differ from MS39p2w174 sequence are shown, red: residues that closely interact with EBNA1AA386-405 according to crystal structure. dots: gaps introduced during IMGT GapAlign for alignment and numbering purposes, numbers: residue numbers according to IMGT unique numbering. Intracellular domain, ICD; extracellular domain, ECD; heavy chain, HC; light chain, LC; complementarity determining region, CDR; framework region, FR; germline, GL.
Extended Data Fig. 8 Extended characteristics of GlialCAMAA370-389 and immunofluorescence stainings with MS39p2w174.
a, Phage display PhiP-Seq data, showing alignment of Pro/Arg-rich region and adjacent residues of all phage display peptides enriched above 100 / 105 reads. b, Immunofluorescence of mouse brain slices stained with (i) control antibody, and (ii-iv) MS39p2w174 (green) and DAPI (blue). (i,ii) full brain, scale bars: 2000 µm, (iii) magnification of hippocampus with prominent MS39p2w174 staining, scale bar: 400 µm, and (iv) olfactory bulb with prominent MS39p2w174 staining in the olfactory nerve (oln), glomerular (gl), and external plexiform layers (epl), but not the mitral (ml), internal plexiform (ipl), or granule cell (gcl) layers, scale bar: 100 µm. c, Immunofluorescence of primary rat oligodendrocytes with isotype control antibody (top panel) and MS39p2w174 (bottom panel). d, K562 cells in culture, wildtype (left) and transduced with full-length GlialCAM (right). e, Immunofluorescence with MS39p2w174 on WT K562 cells (top) and GlialCAM-tg K562 cells (center and bottom). White arrow: single K562 cell, orange arrow: high intensity MS39p2w174 staining on the cell border between transgenic K562 cells in bulks. c, e, Scale bars: 40 µm. b–e, representative micrographs of at least two experiments. f, Overview of phosphorylated residues in GlialCAM, identified by mass spectrometry (phosphoSite.org). The two phosphorylated serine residues of interest are indicated with arrows. g, ELISA, measuring binding of MS39p2w174 to native and citrullinated GlialCAMAA370-389 peptides, means of n = 2 independent experiments, each carried out in triplicates. Wildtype, WT; extracellular domain, ECD; intracellular domain, ICD; phosphorylated serine, pSer; citrulline residue, Cit.
Extended Data Fig. 9 Plasma reactivity against EBNA1 and GlialCAM proteins and peptides in healthy control individuals and MS patients.
a, ELISA measurement of antigen-specific IgG reactivity against peptides EBNA1AA386-405, GlialCAMAA370-389, phosphorylated GlialCAMAA370-389 pSer376, 2x-phosphorylated GlialCAMAA370-389 pSer376 pSer377, and scrambled peptide control in plasma samples of healthy control individuals (n = 50) and MS patients (n = 71). Means ± SD in each patient group is shown. Representative OD (450 nm) measurements of two independent experiments, each carried out in duplicates. **P < 0.01, ***P < 0.001 according to two-tailed Mann-Whitney test, Tukey corrected for multiple comparisons. b, ELISA measurements of antigen-specific IgG reactivity against GlialCAM full-length protein, GlialCAMAA370-389, and phosphorylated GlialCAMAA370-389 pSer376 in plasma samples of a separate cohort of healthy control individuals (n = 31) and MS patients (n = 67). Means ± SD across patient groups are shown. Representative OD (450 nm) measurements of two independent experiments, each carried out in duplicates. *P < 0.05, **P < 0.01 according to two-tailed Mann-Whitney test, Tukey corrected for multiple comparisons. c, ELISA measurements of mAB MS39p2w174 binding to EBNA1AA386-405, without interference as well as blocked with scrambled peptide control, EBNA1AA386-405, and GlialCAMAA370-389 pSer376, as a positive control to (Fig. 3q). Mean OD (450 nm) ± SD of quadruplicate measurements from n = 1 experiment are shown. *P < 0.05, **P < 0.01, ***P < 0.001 according to one-way ANOVA, Tukey corrected for multiple comparisons.
Extended Data Fig. 10 T cell response against EBNAAA386-405.
a, ELISA data showing mouse plasma IgG responses against PLPAA139-151 at the indicated timepoints pre and post EAE induction, for scrambled peptide immunized mice (blue, n = 10) and EBNA1AA386-405 immunized mice (red, n = 10). Mean OD (450 nm) fold change ± SD, significance levels according to unpaired two-tailed Mann-Whitney test. Means ± SD, representative of three independent experiments, each carried out as triplicate measurements. b, T cell proliferation measurement by 3H-thymidine incorporation in splenocytes and lymph node cells of mice immunized with scrambled peptide (blue) and EBNA1AA386-405 (red). Cells from n = 10 mice per group were pooled and mean counts per minute (cpm) ± SD of triplicate measurements are shown. P = 8.9 x 10−5, unpaired two-tailed Student’s t-test, Holm-Sidak corrected for multiple comparisons. c–h, ELISA measurements of cytokines in cell culture supernatant of mouse splenocytes and lymph node cells of mice immunized with scrambled peptide (blue) or EBNA1AA386-405 (red) and re-stimulated with the indicated peptides. Cells from n = 10 mice per group were pooled and mean cpm ± SD of six replicate measurements are shown. c, IFN-γ, d, TNF, e, IL-12, f, IL-10, g, IL-6, h, IL-17, *P < 0.05, significance levels according to unpaired two-tailed Mann-Whitney test, Holm-Sidak corrected for multiple comparisons. i, Representative Luxol Fast Blue stained spinal cords from scrambled peptide group (top panel) and EBNA1AA386-405 group (bottom panel). Scale bars left images: 200 μm, right images: 50 μm. j, Statistical evaluation of Luxol Fast Blue scores, means of at least 4 coronal spinal cord sections per mouse and means ± SD for each group (n = 9) are shown. ****P < 0.0001, unpaired two-tailed Mann-Whitney test. k, l, Flow cytometry data of PBMC from healthy control individuals (n = 6, blue) and MS patients (n = 7, red), showing percent of k, IFN-γ+ and l, IL-17+ CD4+ T cells in all CD4+ T cells. Mean MFI ± SEM are shown for the respective groups. Significance levels were assessed by two-way ANOVA, followed by FDR calculation using the two-stage step-up method of Benjamini, Krieger and Yekutieli, *Significat at FDR < 0.1. m, Flow cytometry data, representative dot plots are shown for two individuals from the data set presented in Fig. 4f. Healthy control individual (left) and MS patient MS16 (right). Expression levels of Granzyme-B (GZMB) and IFN-γ are presented under the indicated stimulations.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1–6, Supplementary Figs. 1 and 2, Supplementary Discussion and additional references.
Source data
Rights and permissions
About this article

Cite this article
Lanz, T.V., Brewer, R.C., Ho, P.P. et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 603, 321–327 (2022). https://doi.org/10.1038/s41586-022-04432-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04432-7
This article is cited by
-
The immunopathogenesis of narcolepsy type 1
Nature Reviews Immunology (2024)
-
Navigating Epstein–Barr virus autoimmunity: role of NK cells and T cells in multiple sclerosis
Signal Transduction and Targeted Therapy (2024)
-
Microbial infection promotes amyloid pathology in a mouse model of Alzheimer’s disease via modulating γ-secretase
Molecular Psychiatry (2024)
-
Autoimmune, Autoinflammatory Disease and Cutaneous Malignancy Associations with Hidradenitis Suppurativa: A Cross-Sectional Study
American Journal of Clinical Dermatology (2024)
-
The elusive nature of the oligoclonal bands in multiple sclerosis
Journal of Neurology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
